vision-research.eu - The Gateway to European Vision Research
![]()

![]()
You are here: vision-research.eu » Vision Research » Vision in the European Focus » 2017 » Identification of gene defects in the Near East
Identification of gene defects leading to non-progressive and progressive ocular diseases in the Near East

 |
Dr. Said El Shamieh studies patient cohorts to identify gene defects underlying different forms of inherited retinal diseases in order to allow them to get enrolled in current clinical trials.
Retinitis Pigmentosa (RP). RP represents the larger group within Inherited Retinal Diseases (IRDs) [1]. Population-based studies showed that 1 in 4,000 individual is word-widely affected [1]. Individuals diagnosed with RP initially complain of night blindness due to rod dysfunction followed by progressive visual field constriction, abnormal color vision, and eventually loss of central vision due to cone photoreceptor involvement [1,2]. RP is inherited as a Mendelian trait in most cases [3]. On the basis of its mode of inheritance and prevalence, RP can be divided into 3 groups: autosomal dominant (ad) (30–40%), autosomal recessive (ar) (50–60%), and Xlinked (xl) (5–15%) [3]. To date, mutations in 60 genes were reported to cause nonsyndromic RP (till January 2017, https://sph.uth.edu/retnet/).
A better characterization of the underlying genetic mechanisms is warranted not only for better patient management and counseling but also to elaborate therapeutic strategies. Mutation detection by Sanger sequencing and microarray analysis are frequently used to identify known and novel gene defects but these methods are time consuming and expensive. Since 2005, next-generation sequencing (NGS) platforms have become widely available in developed countries, reducing the cost of DNA sequencing. NGS was initially designed for targeted gene sequencing involving limited gene numbers. Thus, targeted gene panels are ideal for analyzing specific mutations or genes that have suspected associations with disease [4]. By focusing on genes that are likely to be involved with disease, we reduce expense and focus sequencing resources on your targeted region. However, recent advances has made it possible to sequence cost-effectively nearly all of the coding variation present in an individual human genome, a process termed ‘whole exome sequencing’ (WES) [5,6]. WES has become a powerful new approach for identifying genes that underlie Mendelian disorders in circumstances in which previously reported genes do not show any causal mutation [5,6].
According to studies conducted at the Audo-Zeitz team (Institut de la Vision, Paris, France) and others, a significant proportion of patients do not appear to have mutations in these implicated genes, suggesting novel gene defects yet to be discovered. Therefore, sequencing of the genomic DNA in patients with RP should not only be focused on the above-mentioned genes, but on other genes that might be also implicated. Using the previously described method at the Audo-Zeitz team, we were able to identify novel gene entitled KIZ, which encodes centrosomal protein kizuna to be implicated in RP and more specifically Rod Cone Dystrophy [5].
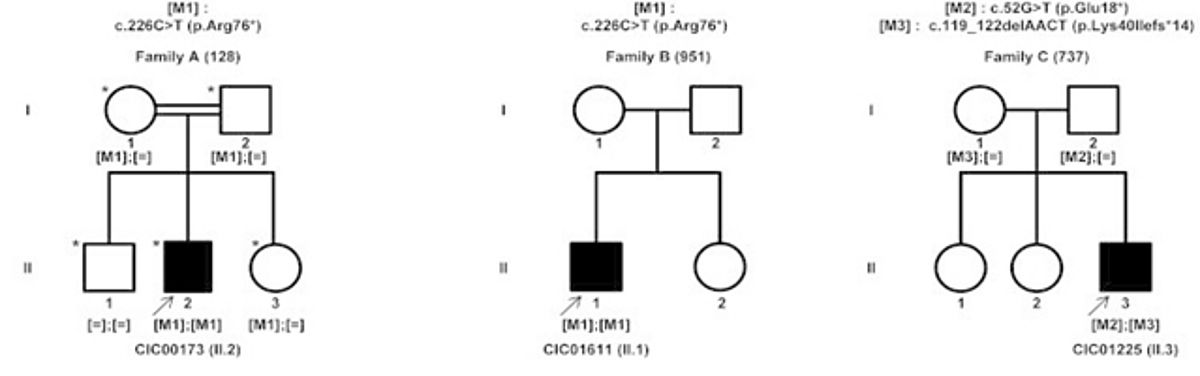 |
Transcriptomic analysis in mice detected mRNA levels of the mouse ortholog (Plk1s1) in rod photoreceptors, as well as its decreased expression when photoreceptors degenerated in rd1 mice. The presence of the human KIZ transcript was confirmed by quantitative RT-PCR in the retina, the retinal pigment epithelium, fibroblasts, and whole-blood cells (highest expression was in the retina). RNA in situ hybridization demonstrated the presence of Plk1s1 mRNA in the outer nuclear layer of the mouse retina.
 |
Since 2015, upon my return home, I have been applying the same approach on Lebanese and middle eastern individuals affected with IRDs. I firmly believe that my current research (done in collaboration with the Audo-Zeitz team) will benefit every affected individual, since identifying the genetic mutations will allow them to get enrolled in current clinical trials.
References
- D. T. Hartong, et al, “Retinitis pigmentosa,” The Lancet, vol. 368, no. 9549, pp. 1795–1809, 2006.
- I. Audo, et al., “Novel C2orf71 mutations account for approximately ∼1% of cases in a large French arRP cohort,” Human Mutation, vol. 32, no. 4, pp. E2091– E2103, 2011.
- A. Anasagasti, et al. “Current mutation discovery approaches in Retinitis Pigmentosa,” Vision Research, vol. 75, pp. 117–129, 2012.
- S. El Shamieh, et al., “Targeted next generation sequencing identifies novel mutations in RP1 as a relatively common cause of autosomal recessive rod-cone dystrophy,” Biomed Res Int. 2015:485624, 2015.
- S. El Shamieh, et al., “Whole-exome sequencing identifies KIZ as a ciliary gene associated with autosomal-recessive rod-cone dystrophy,” American Journal of Human Genetics, vol. 94, no. 4, pp. 625–633, 2014.
- Audo, I., et al. (2014). The familial dementia gene revisited: a missense mutation revealed by whole-exome sequencing identifies ITM2B as a candidate gene underlying a novel autosomal dominant retinal dystrophy in a large family. Hum. Mol. Genet. 23, 491–501, 2014
Contact
Said El Shamieh PhD.
Assistant Prof. Genetics & Genomics
Faculty of Health Sciences
P.O. Box: 11 5020 Beirut, Lebanon
Phone: +961 1 300110 Ext: 2721
Fax: +961 1 818402 Ext: 2589
E-mail: s.elshamieh[at]bau.edu.lb