vision-research.eu - The Gateway to European Vision Research
![]()

![]()
You are here: vision-research.eu » Vision Research » The Young Researchers View » Pietro Farinelli (Q03-2015)
The Research of Pietro Farinelli
 |
Primary cilia are surface-exposed sensory organelles projecting from nearly every quiescent cell type in our bodies. They are composed of a microtubule scaffold called the axoneme, which emanates from a specialized centriole called the basal body. Being at the interface between the extracellular and the intracellular environments, primary cilia play a key role in a wealth of basic cellular functions such as the detection, transmission and coordination of a variety of optical, osmotic, mechanical and chemical signals. In this way, primary cilia coordinate fundamental cellular processes during embryonic development as well as in adult life (Satir et al., 2010).
The outer segment of retinal photoreceptors is considered to be a primary cilium (Fig. 1), which is modified in order to maximize light detection. Similarly to other primary cilia, the photoreceptor cilium does not have its own translational machinery; therefore it needs to import and utilize all the components required to perform normal cellular tasks from the protein synthesis apparatus of the cell body. In photoreceptors, proteins of the outer segment are biosynthesized in the endoplasmic reticulum and Golgi apparatus. Subsequently, they are delivered through motor protein-driven vesicles to the basal body and finally to the outer segment (Wang and Deretic, 2014). The presence of primary cilia in nearly every cell of the human body, together with the crucial functions they perform, implies that abnormalities of the basal body/cilium might be linked with severe diseases collectively called the ciliopathies (Hildebrandt et al., 2011). These include a broad spectrum of disorders affecting tissues and organs such as the kidneys, skin, brain, bones and retina, with the latter tissue being one of the major target of ciliopathies (Wheway et al., 2014).
 |
My main interest at the Department of Medical Genetics at the University of Lausanne is the functional characterization of FAM161A, a putative ciliary protein of still unknown function. The story of FAM161A began during 2010 when our group and others, independently, found that FAM161A is frequently mutated in cases of retinitis pigmentosa, a group of diseases that provokes a slow but irreversible loss of vision, and represents the leading cause of inherited blindness in the developed world. All the mutations identified in FAM161A so far lead to premature stop codons and therefore the absence of the protein. FAM161A contains three coiled-coil domains, two of which are part of the C-terminal UPF0564 domain, a well-conserved structure that mediates protein-protein interactions. FAM161A localizes at the connecting cilium and basal body of both rodent and human photoreceptors (Fig. 2) (Di Gioia et al., 2012). In addition, FAM161A interacts with other proteins involved in retinal ciliopathies (Di Gioia et al., 2012), such as CEP290, LCA5, OFD1 and SDCCAG8. Furthermore, a FAM161A gene trap mouse model is available, which displays retinal degeneration and, as with human disease, with no obvious additional phenotype (Karlstetter et al., 2014).
 |
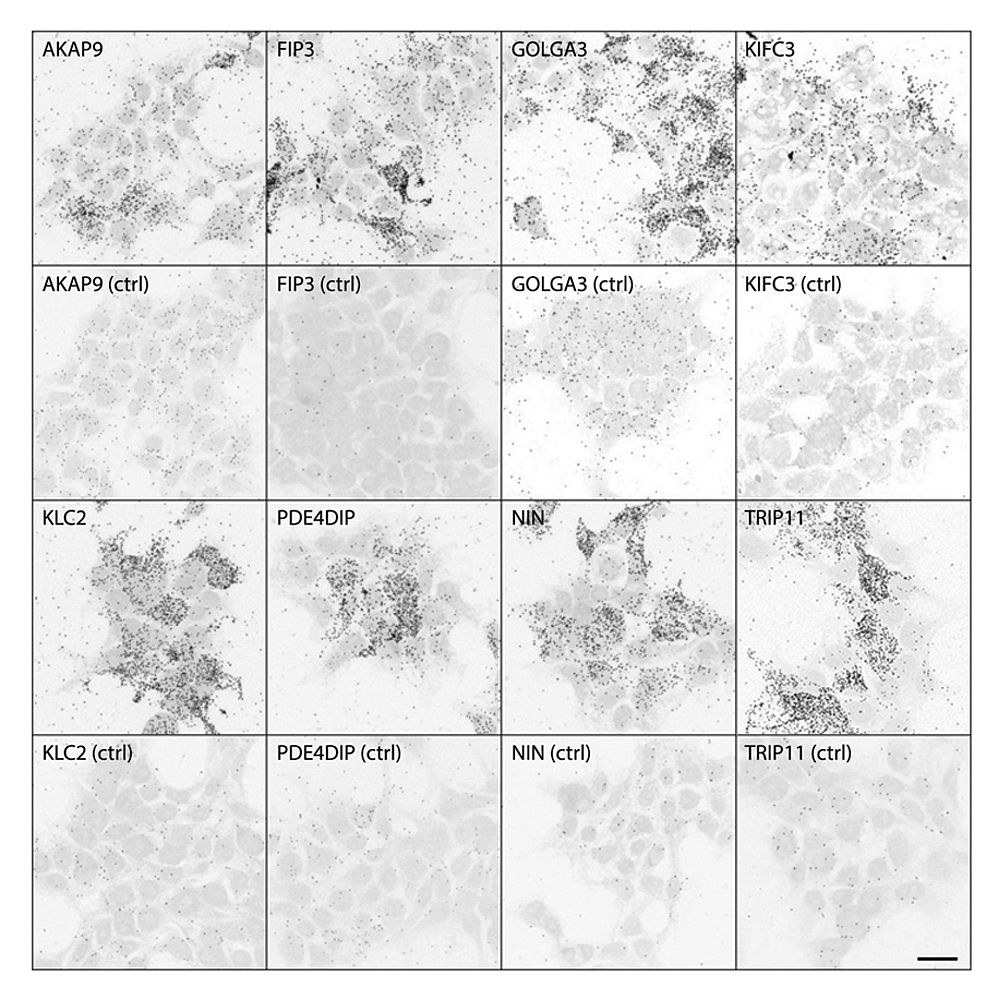 |
Recently, we have unveiled the binary interactome of FAM161A by a GAL4-based yeast two-hybrid (Y2H) screening of bovine and human retinal cDNA libraries. We have in fact identified 53 putative interactors of FAM161A, twenty-three of which were entries in the ciliary proteome database (Gherman et al., 2006). Interestingly, many of the putative interactors identified are associated with specific compartments, notably the Golgi apparatus and the cytoskeleton, and are also associated with intracellular transport as well as centrosomal proteins. This suggests that FAM161A is part of the newly discovered Golgi-centrosome network, a cellular pathway playing a role in ciliogenesis and cellular stability. Using proteomic and immunofluorescence-based tools, such as co-immunoprecipitation and proximity ligation assays on cell lines, we were able to validate eight novel interactors of FAM161A (Fig. 3), namely AKAP9 and PDE4DIP (involved in Golgi and centrosome stability), GOLGA3 and TRIP11 (present at the cis-Golgi and interacting with motor as well as intraflagellar proteins, respectively) FIP3, KIFC3 and KLC2 (proteins involved in the transport of cargo proteins) and NIN (protein of the sub-distal appendages of the mother centriole). These findings indicate that FAM161A is at the heart of a complex protein network and that its role is unlikely to be limited only to ciliary tasks, but also to extend to more general cellular functions, highlighting the existence of novel putative molecular mechanisms triggering the pathology of retinal disease.
Future work is needed to elucidate the aetiology behind the detrimental impact of the lack of FAM161A in the retina, but not in other tissues where FAM161A is also expressed (like testis and kidney). Worth noting is that FAM161B, the orthologue of FAM161A, is not currently associated with disease. Therefore, it would be interesting to clarify whether FAM161B, functions in parallel and/or redundantly with FAM161A in controlling primary cilia and centrosome physiology.
References
- Di Gioia, S.A., Letteboer, S.J.F., Kostic, C., Bandah-Rozenfeld, D., Hetterschijt, L., Sharon, D., Arsenijevic, Y., Roepman, R., and Rivolta, C. (2012). FAM161A, associated with retinitis pigmentosa, is a component of the cilia-basal body complex and interacts with proteins involved in ciliopathies. Human Molecular Genetics 21, 5174-5184.
- Gherman, A., Davis, E.E., and Katsanis, N. (2006). The ciliary proteome database: an integrated community resource for the genetic and functional dissection of cilia. Nat Genet 38, 961-962.
- Hildebrandt, F., Benzing, T., and Katsanis, N. (2011). Ciliopathies. New England Journal of Medicine 364, 1533-1543.
- Rachel, R.A., Li, T., Swaroop, A. (2012) Photoreceptor sensory cilia and ciliopathies: focus on CEP290, RPGR and their interacting proteins. Cilia. Dec 3;1(1):22.
- Karlstetter, M., Sorusch, N., Caramoy, A., Dannhausen, K., Aslanidis, A., Fauser, S., Boesl, M.R., Nagel-Wolfrum, K., Tamm, E.R., Jägle, H., et al. (2014). Disruption of the retinitis pigmentosa 28 gene Fam161a in mice affects photoreceptor ciliary structure and leads to progressive retinal degeneration. Human Molecular Genetics 23, 5197-5210.
- Satir, P., Pedersen, L.B., and Christensen, S.T. (2010). The primary cilium at a glance. Journal of Cell Science 123, 499-503.
- Wang, J., and Deretic, D. (2014). Molecular complexes that direct rhodopsin transport to primary cilia. Progress in Retinal and Eye Research 38, 1-19.
- Wheway, G., Parry, D.A., and Johnson, C.A. (2014). The role of primary cilia in the development and disease of the retina. Organogenesis 10, 69-85.
Pietro Farinelli Ph.D
Phone: +41 (21) 692 54 70
Website: unil.ch/dgm/home/menuinst/research-groups/carlo-rivolta.html
Contact
University of Lausanne
Faculty of Biology and Medicine
Department of Medical Genetics
Rue du Bugnon 27
CH-1005 Lausanne
Switzerland