vision-research.eu - The Gateway to European Vision Research
![]()

![]()
You are here: vision-research.eu » Vision Research » The Young Researchers View » Susanne Roosing (Q01-2019)
The Research of Susanne Roosing
 |
Background
My research focused on non-syndromic retinal dystrophy and later on syndromic disease containing retinal dystrophy in its clinical spectrum. The aim of my PhD project in the lab of Prof. Frans P.M. Cremers in Nijmegen was to elucidate novel genetic causes for retinal dystrophies using homozygosity mapping and WES, to increase the molecular genetic knowledge of inherited retinal dystrophies (IRDs).[1]
After my PhD training, I transitioned for a 2-year postdoctoral training to the laboratory of Prof. Joseph G. Gleeson who specializes in exploring mechanisms causative for pediatric brain diseases at the Rockefeller University, New York, USA. My project was directed to study cilia in a syndromic disorder, Joubert syndrome with retinal dystrophy in its disease spectrum. Amongst others, this study has resulted a publicly available dataset (coined CILIOGENESIS) which presents a list of candidates for involvement in ciliary processes based on a machine learning approach on whole genome siRNA study performed in a dual fluorescent cell-line. This dataset enabled the identification of the gene KIAA0586 which when mutated is causative for Joubert syndrome.[2] I returned to the Department of Human Genetics in Nijmegen, the Netherlands, in June 2016 were I am developing my research niche in order to identify and interpret the missing mutations in inherited retinal dystrophies (IRDs) through whole genome sequencing efforts.
Research
The leading causes of hereditary blindness worldwide are retinal disorders, which primarily affect the photoreceptors. Thus far, genes causing inherited retinal dystrophies (IRDs) were found in major pathways of the visual system like the phototransduction cascade, retinoid cycle, transport processes, disk membrane morphogenesis, and synaptic transduction. Although important steps are being made, there are currently limited therapeutic options to inhibit, stop, or reverse the degenerative disease process. In order to develop effective therapeutic approaches the genetic etiology of IRDs needs to be elucidated. Over 260 genes have been found to cause IRD, but mutations in these genes explain only 60-80% of all IRDs, leaving many cases without a genetic diagnosis.
The large number of genes underlying IRDs illustrates the high genetic heterogeneity, and the number of disease genes is anticipated to increase when taking into account all currently unsolved cases (Figure 1). While in the early days of genetic studies findings were made relatively easy with multiple families with mutations in the same gene, now this low-hanging fruit is gone. Where in the past several families could carry pathogenic variants in the same gene, now the era has come where usually the supporting evidence is only a single family is found with pathogenic variants in a candidate gene.
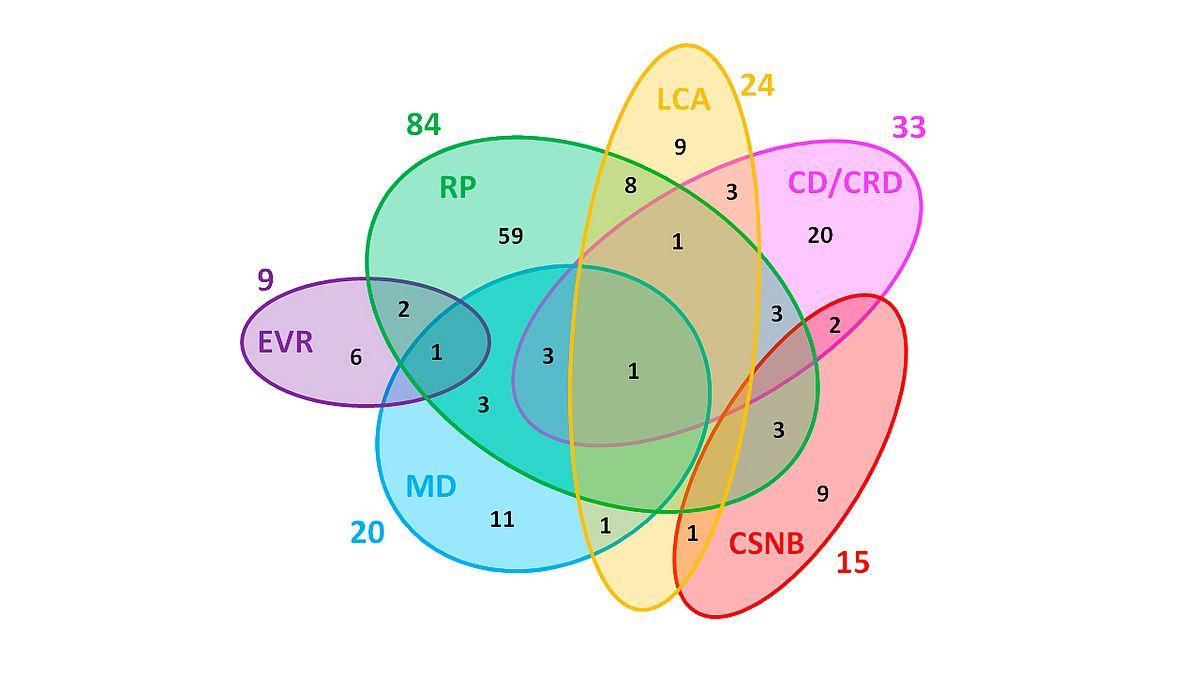 |
Current studies, in most cases, are based on whole exome sequencing (WES) as it is most time- and cost-effective and studies all coding parts of the genome in a single experiment. However, often it is not easy to pinpoint the genetic defect(s) among the ~60.000 variants that are identified by WES. In the past years, we have learned several lessons from the gene identification studies.
Upcoming gene identification approaches should thus take all possible genes and variants into consideration. In previous studies, we have shown that well established syndromic associated genes such as MFSD8 leading to early lethality could also give rise to IRD only due to a different mutational mechanism.[3] Visual impairment is the first symptom in the majority of these syndromes, but as the disease progresses over time and other features occur, minor attention is assigned to the retinal phenotype. Second, pathogenic variants may have an unexpected effect as we showed that CEP290 nonsense mutations may also have an effect on splicing, which lead to the skipping of exons carrying the truncating mutation.[4] More recent studies are showing the relevance of non-canonical or deep intronic variants affecting the process of splicing by creating a strong acceptor or donor splice site or disrupting an exonic splice enhancer or silencer. The potential effect of all types of variants on splicing should thus be taken into account when analyzing WES or WGS data. (Verbakel in preparation, Fadaie in preparation)
Collaboration
Whole genome sequencing (WGS) is gradually entering routine diagnostics, but the interpretation of ~6 billion non-coding variants is very challenging without proper functional read-outs. We will employ WGS in selected IRD cases that could not be solved using WES analysis and for which we previously obtained positional information through homozygosity or linkage mapping. My aim is to elucidate novel genetic causes for IRD using whole genome sequencing (supported by homozygosity mapping, linkage regions and/or pathogenicity clues gained from prior studies), to increase the molecular genetic knowledge of IRDs and open new avenues for generating therapeutic approaches for all patients.
While aiming to uncover novel strategies to finding new causative genes for IRD, this cannot be pursued without extensive collaboration with other research groups in Europe and the world. The urgency for collaborations has becoming more clear as individual research groups have interesting candidate genes which may explain IRD in single families. The European Retinal Disease Consortium (ERDC; www.erdc.info) has a long-standing and ongoing collaboration in sharing patient resources, data and candidate genes as well as technologies. Since early 2017, I am the coordinator where today, the ERDC contains 22 research groups of which 20 are European based and two are American and Canadian (Figure 2). This collaboration has resulted in over 140 joint publications and many joint projects, where the aim is to provide all IRD patients with a genetic diagnosis.
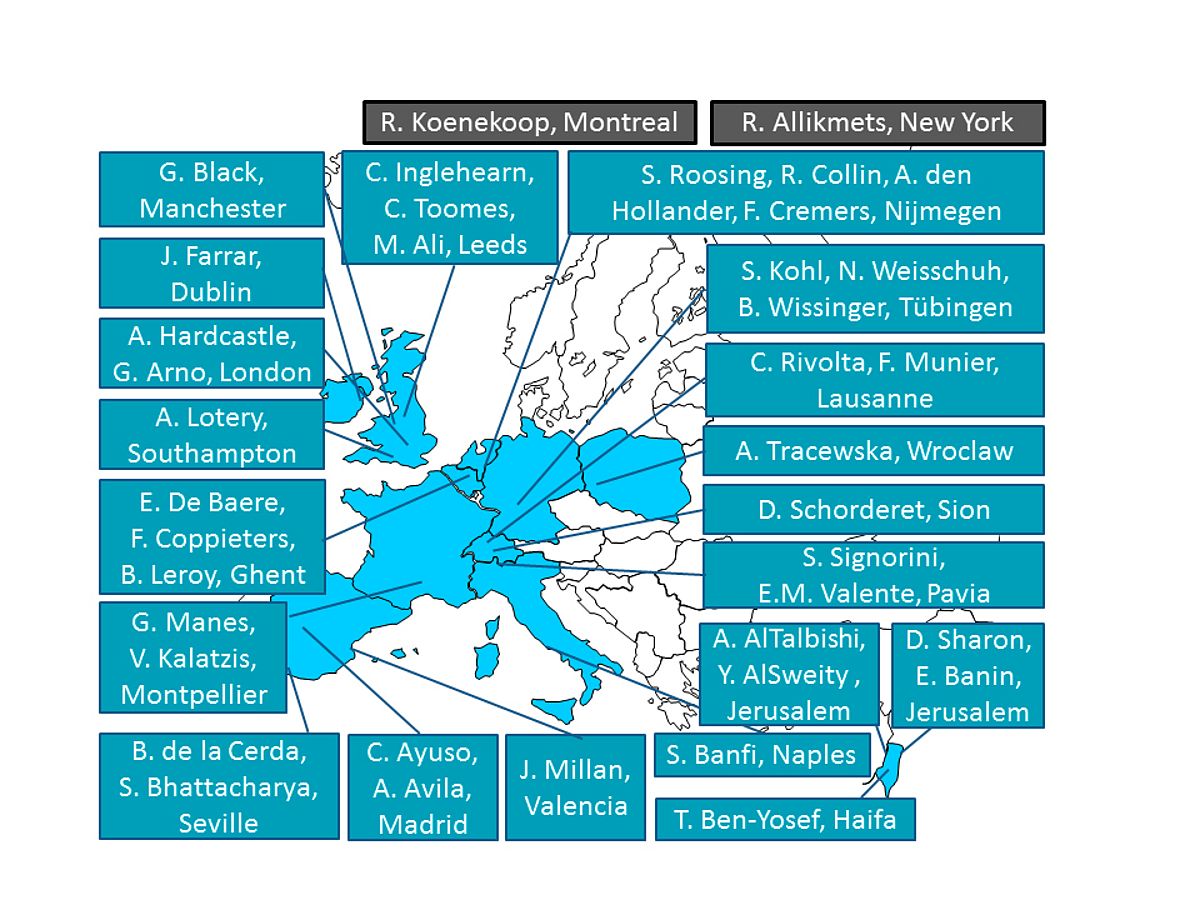 |
Our publication ‘Identification of Inherited Retinal Disease-Associated Genetic Variants in 11 Candidate Genes’ would not have been possible without the collaboration of the European research groups and data-sharing.[5] Strong candidate genes were presented to the scientific community as we have not been able to spot additional families with mutations in these genes even when the consortium has joint forces.
This collaborative research will directly benefit individuals with IRDs and their families through improved diagnostic, prognostic and recurrence risk information as novel causative genes will be identified. Moreover, the elucidation of the causative defect in these IRD patients will improve monitoring and will provide early treatment for medical complications
References
- Roosing S, Thiadens AA, Hoyng CB, Klaver CC, den Hollander AI, Cremers FP (2014) Causes and consequences of inherited cone disorders. Prog Retin Eye Res (0). doi:10.1016/j.preteyeres.2014.05.001
- Roosing S, Hofree M, Kim S, Scott E, Copeland B, Romani M, Silhavy JL, Rosti RO, Schroth J, Mazza T, Miccinilli E, Zaki MS, Swoboda KJ, Milisa-Drautz J, Dobyns WB, Mikati MA, Incecik F, Azam M, Borgatti R, Romaniello R, Boustany RM, Clericuzio CL, D'Arrigo S, Stromme P, Boltshauser E, Stanzial F, Mirabelli-Badenier M, Moroni I, Bertini E, Emma F, Steinlin M, Hildebrandt F, Johnson CA, Freilinger M, Vaux KK, Gabriel SB, Aza-Blanc P, Heynen-Genel S, Ideker T, Dynlacht BD, Lee JE, Valente EM, Kim J, Gleeson JG (2015) Functional genome-wide siRNA screen identifies KIAA0586 as mutated in Joubert syndrome. Elife 4. doi:10.7554/eLife.06602
- Roosing S, van den Born LI, Sangermano R, Banfi S, Koenekoop RK, Zonneveld-Vrieling MN, Klaver CC, van Lith-Verhoeven JJ, Cremers FP, den Hollander AI, Hoyng CB (2015) Mutations in MFSD8, encoding a lysosomal membrane protein, are associated with nonsyndromic autosomal recessive macular dystrophy. Ophthalmology 122 (1):170-179. doi:10.1016/j.ophtha.2014.07.040
- Roosing S, Cremers FPM, Riemslag FCC, Zonneveld-Vrieling MN, Talsma HE, Klessens-Godfroy FJM, den Hollander AI, van den Born LI (2017) A Rare Form of Retinal Dystrophy Caused by Hypomorphic Nonsense Mutations in CEP290. Genes 8 (8). doi:10.3390/genes8080208
- Astuti GDN, van den Born LI, Khan MI, Hamel CP, Bocquet B, Manes G, Quinodoz M, Ali M, Toomes C, McKibbin M, El-Asrag ME, Haer-Wigman L, Inglehearn CF, Black GCM, Hoyng CB, Cremers FPM, Roosing S (2018) Identification of Inherited Retinal Disease-Associated Genetic Variants in 11 Candidate Genes. Genes 9 (1). doi:10.3390/genes9010021
- Khan M, Fadaie Z, Cornelis SS, Cremers FPM, Roosing S (2019) Identification and Analysis of Genes Associated with Inherited Retinal Diseases. Methods in molecular biology (Clifton, NJ) 1834:3-27. doi:10.1007/978-1-4939-8669-9_1

Susanne Roosing, PhD
Postdoctoral researcher Blindness Genetics
Radboud University Medical Center
Department of Human Genetics (route 855)
Geert Grooteplein-Zuid 10
6525 GA Nijmegen
The Netherlands
E-mail: Susanne.Roosing[at]radboudumc.nl
Phone: +31 24 3668901
Postbus 9101, 6500 HB Nijmegen (855)
Website: http://www.ru.nl/donders/research/theme-2-perception/blindness-genetics