vision-research.eu - The Gateway to European Vision Research
![]()

![]()
You are here: vision-research.eu » Vision Research » The Young Researchers View » Karin Littink (Q02-2010)
Elucidation of genetic causes of retinal dystrophies using homozygosity mapping.

Purpose of finding genetic cause of retinal dystrophies
Since two decades, the genetic causes of retinal dystrophies are being elucidated, which provided insights into the normal visual processes, disease etiology, and genetic mechanisms leading to a disease, which in turn can lead to new therapeutic options. In addition, the patient can benefit from knowing the genetic cause, since it may give more specific knowledge about the mode of inheritance, the prognosis, and dietary advice to take or to avoid additional vitamin A. These studies have also led to the exciting development of a gene-specific therapy for patients carrying mutations in RPE65.1, 2
With respect to the genetically heterogeneous retinal diseases, the genetic defects can now be detected in ~50% of autosomal recessive or autosomal dominant retinitis pigmentosa (RP) patients, ~70% of Leber congenital amaurosis (LCA), ~45% of autosomal recessive cone-rod dystrophy (CRD), ~10% of autosomal recessive cone dystrophy and ~80% of autosomal recessive achromatopia patients.
Homozygosity mapping approach
We use a homozygosity mapping approach to detect new mutations. This method is regularly used in mutation detection in consanguineous families – i.e. families in which the parents of a patient are related to each other. Figure 1 shows how a mutation may be transferred through several generations, and finally is present in a patient. The homozygous regions surrounding the mutation can be detected as a stretch of homozygous single nucleotide polymorphisms (SNPs) in genome-wide SNP array analysis.
Actually, homozygous mutations were found in about 35% of patients with retinal dystrophy from non-consanguineous families as well. This led to the hypothesis that also in non-consanguineous families, the parents of a patients may have a common ancestor who passed on the mutation (Figure 2), for example in regions where people have the tendency to stay in the same small village, and marry with a person from the same village. Therefore, we analysed DNA of 95 CRD patients using a whole-genome SNP-array and performed homozygosity mapping using Plink software.
Major results up to now
The search for overlap of homozygous regions with known CRD genes (ABCA4, ADAM9, CERKL, PROM1, RPGRIP1) led to the identification of novel mutations in three of these genes (in ABCA4 in 2 patients, CERKL in 2 patient and in PROM1 in 1 patient).3 Further screening of known retinal dystrophy genes residing in a homozygous region led to the identification of novel mutations in two genes (CABP4 and KCNV2).3, 4 Clinical re-evaluation of these patients revealed that their phenotypes were not CRD, but cone-related dystrophies as described previously for these genes.
Overlap of a 5Mb homozygous region with the previously described 15Mb RP25 locus led to the identification of a new gene, EYS.5 (simultaneous with the groups of Antinolo (Sevilla) and Bhattacharya (London)6 ). Mutations in EYS appeared to be mostly associated to RP.
In conclusion, homozygosity mapping in mostly non-consanguineous families led to the identification of the causative mutation in 4 out of 11 families, and in four individual patients, up to now. The results show that homozygosity mapping is most effective in families with more than 1 affected sibling, but can lead to the identification of the genetic defect as well in individual patients, especially when the grandparents of maternal and paternal side are from one region.
Future perspectives
For future perspectives, homozygosity mapping will become more powerful in combination with next generation sequencing, a technique that can sequence a huge number of nucleotides in one run.
Personally, I hope that many more genes will be detected, that gene- and/or mutation-specific therapies will become a realistic future perspective for all patients, and that I will be able to stay involved in this kind of research from the clinical side as well, as an ophthalmologist.
Figure 1
Segregation of a haplotype harbouring a mutation in a consanguineous family.
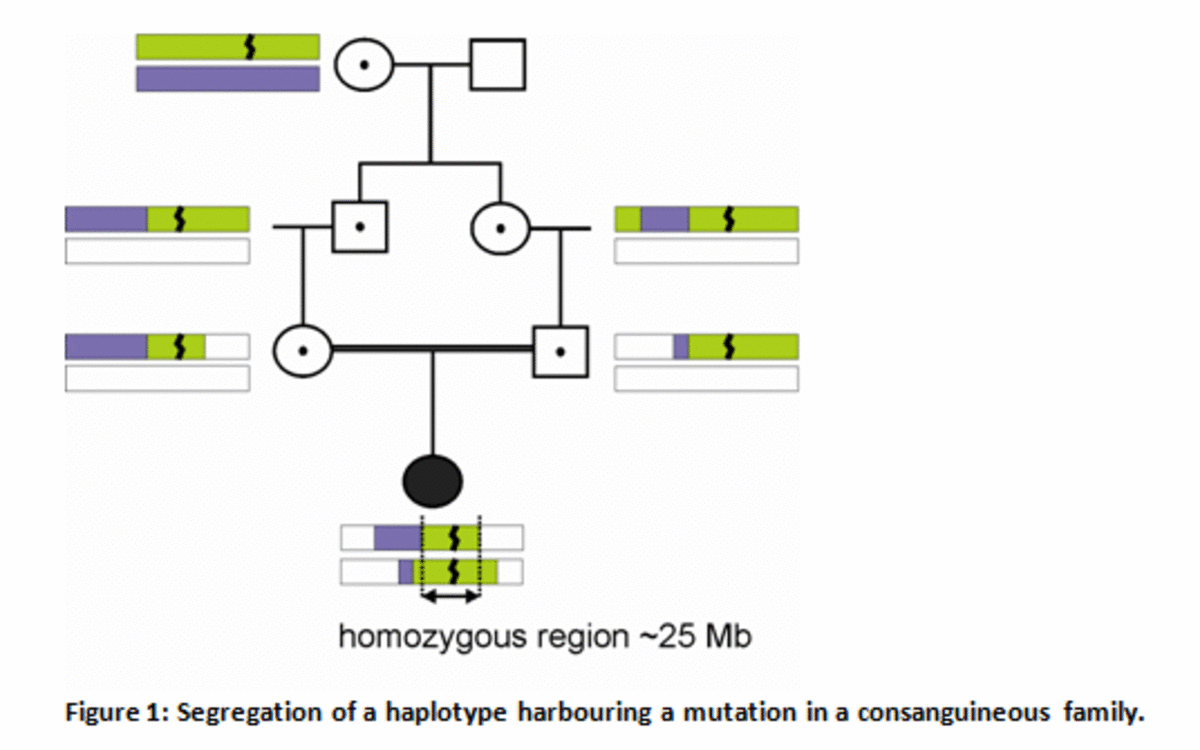
The great-grandmother carries a mutation ( ), that is passed on to the next generations on a haplotype that shortens every generation due to recombination. Via the maternal and paternal side this mutation is then present on both chromosomes in the patient. Because the patient carries the ancestral haplotype on both alleles, the region surrounding the mutation is homozygous. Due to the limited number of generations between the ancestor carrying the mutation and the patient, homozygous regions can reach a length of approximately 25 Mb.
Figure 2
Segregation of haplotype harbouring a mutation in a non-consanguineous family.
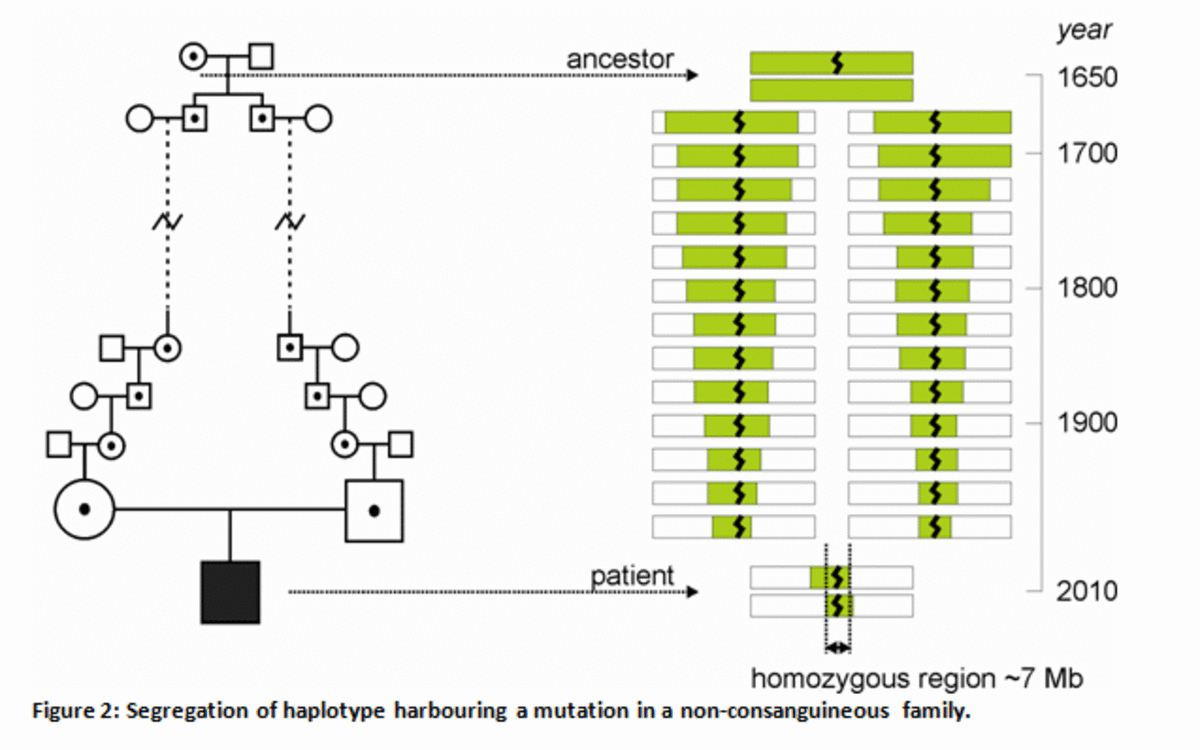
An ancestor carrying a mutation ( ) passed on the mutation including part of the surrounding haplotype to the next generations, and via several generations, the ancestral haplotype is present on both chromosomes of the patient. Due to many recombinations between the ancestor and the patient, homozygous regions in the patient will be relatively small (~7 Mb).
References
- Bainbridge JW, Smith AJ, Barker SS, et al.
Effect of gene therapy on visual function in Leber's congenital amaurosis.
N Engl J Med. 2008;358:2231-2239. - Maguire AM, Simonelli F, Pierce EA, et al.
Safety and efficacy of gene transfer for Leber's congenital amaurosis.
N Engl J Med. 2008;358:2240-2248. - Littink KW, Koenekoop RK, van den Born LI, et al.
Homozygosity mapping in patients with cone-rod dystrophy: novel mutations and reappraisal of the clinical diagnosis.
Invest Ophthalmol Vis Sci. 2010;2010, in preparation. - Littink KW, van Genderen MM, Collin RWJ, et al.
A novel homozygous nonsense mutation in CABP4 causes congenital cone-rod synaptic disorder.
Invest Ophthalmol Vis Sci. 2009;50:2344-2350. - Collin RWJ, Littink KW, Klevering BJ, et al.
Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa.
Am J Hum Genet. 2008;83:594-603. - Abd El-Aziz MM, Barragan I, O'Driscoll CA, et al.
EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa.
Nat Genet. 2008;40:1285-1287.
Karin Littink
Radboud University Nijmegen
Medical Centre
The Rotterdam Eye Hospital