vision-research.eu - The Gateway to European Vision Research
![]()

![]()
You are here: vision-research.eu » Vision Research » The Young Researchers View » Daniele Dell’Orco (Q02-2020)
The Research of Daniele Dell’Orco
 |
Background
I studied Physics and Biophysics at the University of Parma (Italy) and Lund (Sweden) before obtaining a PhD in Biotechnology and Molecular Medicine from the University of Modena and Reggio Emilia (Italy). During my pre-doctoral activity, I visited several laboratories in Sweden and in Germany, then I received an A. von Humboldt Research Fellowship, which allowed me to spend a PostDoc period at the University of Oldenburg (Germany).
In December 2011 I moved to the University of Verona, where I was appointed Assistant Professor. I am currently Associate Professor in the same university, where I am leading a research group at the Biological Chemistry section of the Department of Neurosciences.
I would define my background as truly interdisciplinary. From the very beginning, my research has involved an integration of experimental and computational investigations of complex molecular mechanisms in biology. Research on vision is itself exquisitely multifaceted, and it allows one to tackle the same biological/biomedical process by many different viewpoints. Today, I could not ask for a better framework, in which to continue astonish myself every day.
A bottom-up view of the mechanisms triggering vision
Vision is triggered by the absorption of light in rods and cones. In spite of species-dependent characteristics, there is a common molecular machinery that allows photoreceptors to convert the information carried by light (an electromagnetic entity) into a chemical signal propagating intracellularly, and then again into an electrical information that can travel through an intricate neuronal network eventually resulting in the visual perception. Since the beginning of my career, I have been deeply fascinated by how it is possible to achieve such a temporarily and spatially well-defined signaling cascade known as phototrasduction.
After all, from a very reductionist perspective rhodopsin, trandsducin and ionic channels are proteins; cyclic GMP and calcium are molecules and ions; the hyperpolarization of the membrane that signals the light absorption is nothing but a temporary suppression of the electrical current circulating through the cell membrane.
How can one put together all these individually resolved pieces to get a working puzzle?
By using a bottom-up kinetic modeling approach, that starts by a comprehensive description of each individual interaction, we have been able to put together a sufficiently accurate model of this intricate network of protein-protein, protein-nucleotide, protein-ion interaction that builds up the phototrasduction cascade. The resulting phototransduction model was able to recapitulate the salient features in the slower photoresponse of amphibians [1] and in the faster response of higher vertebrates including mice [2]. This allows to simulate, at molecular detail, the dysregulation of the cascade in disease-associated conditions, and consequently, to predict the effect of specific therapeutic interventions.
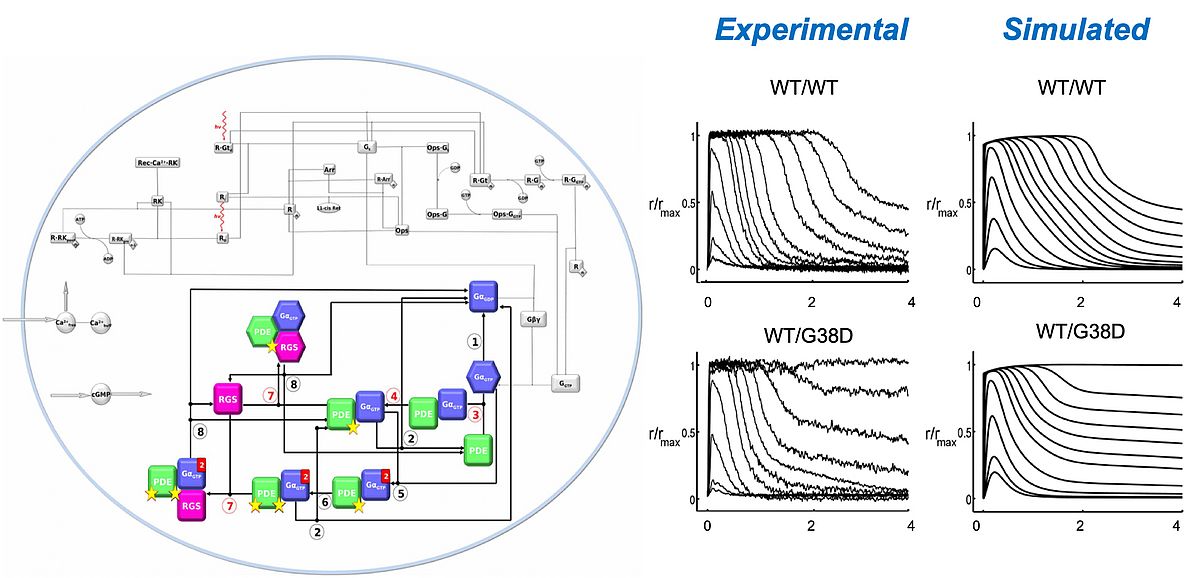 |
The role of calcium sensor proteins in phototransduction and in cone dystrophies
The maintenance of second messenger homeostasis in photoreceptor cells is fundamental for their normal function. Calcium (Ca2+) and cGMP levels are strictly interdependent and are regulated by means of the negative feedback loop acting on the retinal guanylate cyclase (GC1) [4]. GC1 is activated at low Ca2+ by GC-activating proteins, especially GCAP1, that is present both in rods and cones. GCAP1 is a neuronal Ca2+ sensor capable of detecting subtle changes in intracellular Ca2+ by binding the cation into specific structural motifs. In light-activated conditions, the levels of both cGMP and Ca2+ in the outer segment drop and GCAP1 replaces the bound Ca2+ with Mg2+, thus switching to a conformation that is capable of stimulating GC1 and enhance the synthesis of cGMP to restore dark levels. When the phototransduction cascade is switched off, Ca2+ levels return at their dark levels, and Ca-GCAP1 is again ready to inhibit the catalytic activity of GC1.
More than twenty point mutations have been found in GUCA1A, the gene encoding GCAP1, in patients suffering from cone or cone-rod dystrophies (COD/CORD).
We aim at understanding, at the molecular level, the genotype/phenotype correlation that is responsible for cell degeneration. We use a combination of biochemical and biophysical techniques and integrate our results with extensive molecular simulations of the system under the same conditions tested experimentally. This analysis helps us understand the common hallmarks of disease at the protein level, and design therapeutic strategies for these currently incurable diseases.
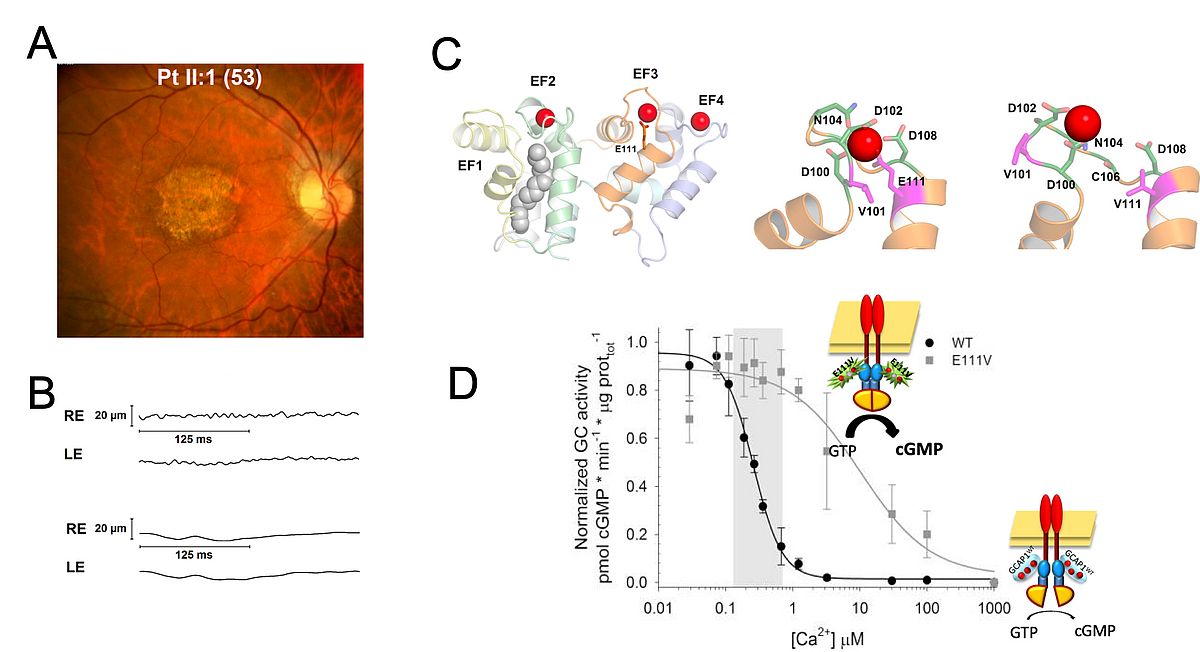 |
A protein-therapy approach to cure retinal degeneration?
Retinal dystrophies associated with GUCA1A have an autosomal dominant inheritance pattern, thus implying that a pool of normal GCAP proteins is present in photoreceptors together with the mutated variant. Our biochemical studies corroborated by the numerical simulation of the phototransduction cascade suggest that increasing the levels of normal (i.e. wild type) GCAP1 in the photoreceptor outer segment would partly attenuate the mutant-induced dysregulation of cGMP signaling and would bring the levels of both cGMP and Ca2+ close to their normal levels [6]. The increase of both cGMP and Ca2+ levels can in fact trigger cell death.
We have promising preliminary data [7] that suggest that liposomes encapsulated by highly pure recombinant proteins can serve as carriers to deliver functional proteins in retinal photoreceptors and modulate their physiological response. Liposomes protect the encapsulated proteins from degradation while constituting biocompatible carriers not triggering inflammation or immune response. Finding an effective way for liposome topical administration could lead to an effective treatment of retinal dystrophies by a novel mutation-independent protein-therapy approach. Our last experimental efforts follow this direction.
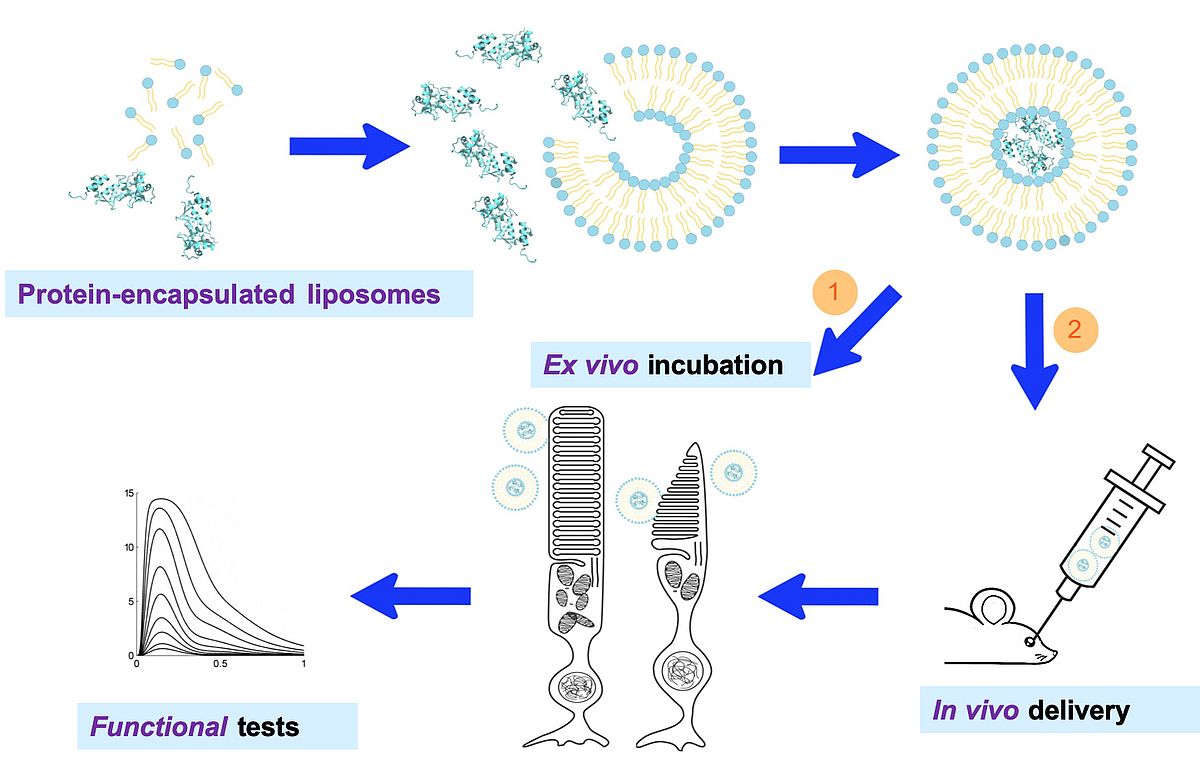 |
References
- Dell'Orco D, Schmidt H, Mariani S, Fanelli F. Network-level analysis of light adaptation in rod cells under normal and altered conditions. Mol Biosyst. 2009;5(10):1232-46. doi: 10.1039/b908123b.
- Invergo BM, Dell'Orco D, Montanucci L, Koch KW, Bertranpetit J. A comprehensive model of the phototransduction cascade in mouse rod cells. Mol Biosyst. 2014 Jun;10(6):1481-9. doi: 10.1039/c3mb70584f.
- Mariani S, Dell'Orco D, Felline A, Raimondi F, Fanelli F. Network and atomistic simulations unveil the structural determinants of mutations linked to retinal diseases. PLoS Comput Biol. 2013;9(8):e1003207. doi: 10.1371/journal.pcbi.
- Koch KW, Dell'Orco D. A calcium-relay mechanism in vertebrate phototransduction. ACS Chem Neurosci. 2013;4(6):909-17. doi: 10.1021/cn400027z
- Marino V, Dal Cortivo G, Oppici E, Maltese PE, D'Esposito F, Manara E, Ziccardi L, Falsini B, Magli A, Bertelli M, Dell'Orco D. A novel p.(Glu111Val) missense mutation in GUCA1A associated with cone-rod dystrophy leads to impaired calcium sensing and perturbed second messenger homeostasis in photoreceptors. Hum Mol Genet. 2018 Dec 15;27(24):4204-4217. doi: 10.1093/hmg/ddy311.
- Dell'Orco D, Dal Cortivo G. Normal GCAPs partly compensate for altered cGMP signaling in retinal dystrophies associated with mutations in GUCA1A. Sci Rep. 2019 Dec 27;9(1):20105. doi: 10.1038/s41598-019-56606-5.
- Asteriti S, Dal Cortivo G, Pontelli V, Cangiano L, Buffelli M, Dell'Orco D. Effective delivery of recombinant proteins to rod photoreceptors via lipid nanovesicles. Biochem Biophys Res Commun. 2015 Jun 12;461(4):665-70. doi: 10.1016/j.bbrc.2015.04.088

Dr. Daniele Dell’Orco
Associate Professor of Biochemistry
University of Verona
Department of Neurosciences, Biomedicine and Movement Sciences
Section of Biological Chemistry
Strada le Grazie 8
37134 Verona
Italy
E-mail: daniele.dellorco[at]univr.it
Tel. +39-045-802-7637