vision-research.eu - The Gateway to European Vision Research
![]()

![]()
You are here: vision-research.eu » Vision Research » Visionary of the Quarter » Frans Cremers (Q03-2017)
Utilizing genomics and transcriptomics to identify the hidden causes of inherited retinal diseases
 |
Historical background
Through various technological improvements the vision research community in the last three decades was able to identify the large majority of genes underlying inherited retinal diseases (IRDs). Between 1987 and 1998, when we were still assembling the human genome using genome walking and jumping techniques, ‘old-school’ positional cloning studies making use of hemizygous microdeletions in affected males, facilitated the identification of the first genes underlying IRDs, i.e. X-linked choroideremia,1 Norrie Disease2 and X-linked retinitis pigmentosa (RP).3,4 Genetic linkage, candidate gene analysis and homozygosity mapping studies thereafter were successfully employed to identify many genes implicated in autosomal recessive and dominant retinal diseases (Figure 1).5-8
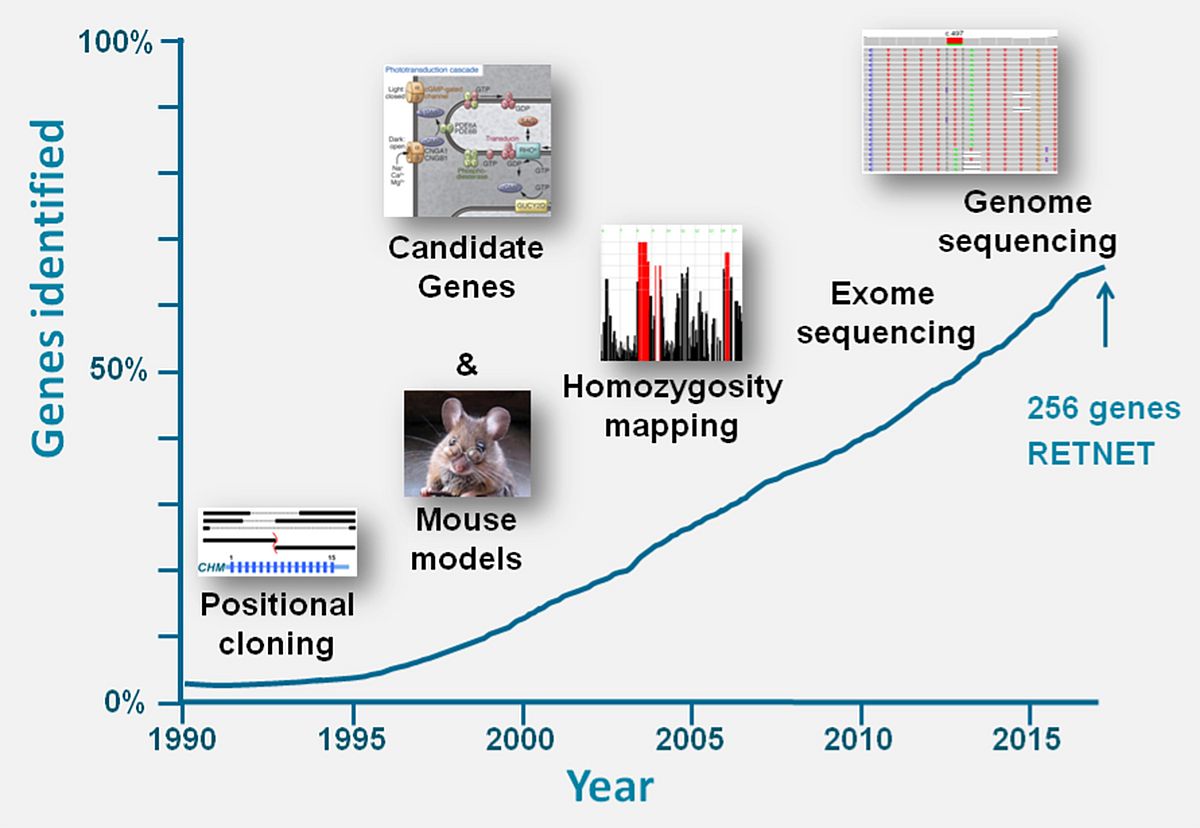 |
In the last decade, whole-exome sequencing (WES) made it possible to identify many more IRD-associated genes as well as genes implicated in familial exudative vitreoretinopathy.9-11 Based on ongoing WES studies (Susanne Roosing et al. unpublished data), in which only a few novel IRD-associated genes are detected, we estimate that more than 80% of the ‘mutational load’ for non-syndromic IRDs has been identified in the currently known ~160 non-syndromic IRD-associated genes (https://sph.uth.edu/retnet/sum-dis.htm#A-genes). Nevertheless, WES analysis on average solves only ~60% of IRDs12 suggesting that a large fraction of variants is still elusive. It is our aim to design novel approaches to identify all the remaining hidden mutations in IRDs in the next decade and to decipher their functional consequences. Below, I describe the new technologies that we have implemented and are developing to reach these goals.
Seeing the complete genomic picture: whole exome sequencing
WES studies are incomplete as the complete enrichment of coding regions, constituting ~2% of the human genome, is technically challenging. GC-poor and GC-rich regions are not or poorly enriched. In addition, many structural variations are missed due to the small sequencing read length in WES. Most importantly, 98% of the genome not carrying coding regions are not investigated for the presence of causal variants. Whole genome sequencing (WGS), and in particular WGS techniques in which no PCR amplification is needed, are superior in terms of sensitivity and completeness. As the costs of WGS have dropped tremendously in recent years, WGS in the Western world will rapidly become the standard technology to identify mutations in human genetic conditions such as IRDs. Long-read sequencing (e.g. PacBio technology) enables the analysis of DNA fragments up to 40 kb in size, with no hindrance of variable GC-content or sizeable repeats, such as LINEs. The generation of more than 6 billion bps of a human genome can now readily be achieved, but the question is how to make sense of the few million small and sizeable variations in each analyzed individual. How can we find the 'needles in the haystacks', i.e. the one or two variants responsible for a monogenic eye condition?
Identifying RNA defects using stem cell technology: ABCA4 and Stargardt disease
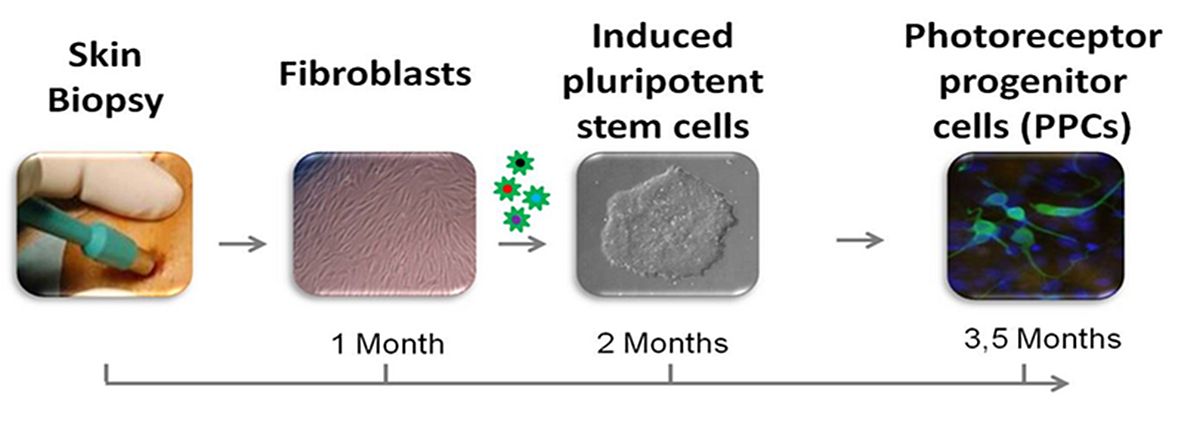
We believe novel read-out systems are needed to make sense of this vast amount of variant data. My team focusses on RNA defects that are the consequences of rare DNA variants. Approximately two-third of known IRD-associated genes is not or poorly expressed in accessible somatic tissues such as skin and blood cells. To bypass this problem, we have taken advantage of one of the most fascinating technological innovations in the last decades, i.e. to reprogram somatic cells into induced pluripotent stem cells (iPSCs), which then can be used to generate photoreceptor progenitor cells (PPCs) in a '2D' configuration or '3D' retinal organoids mimicking the development of an eye-like structure. Using fibroblasts, and more recently peripheral blood cells, we generated iPSCs from patients with Stargardt disease (STGD1) and other IRDs and differentiated these into PPCs (Silvia Albert et al., unpublished data; Figure 2) and retinal organoids. In this way we were able to show the severe effect (exon skipping) of a predicted mild non-canonical splice site mutation in ABCA4 that thereby is the most frequent severe ABCA4 variant in persons with STGD1 or cone-rod dystrophy.13
 |
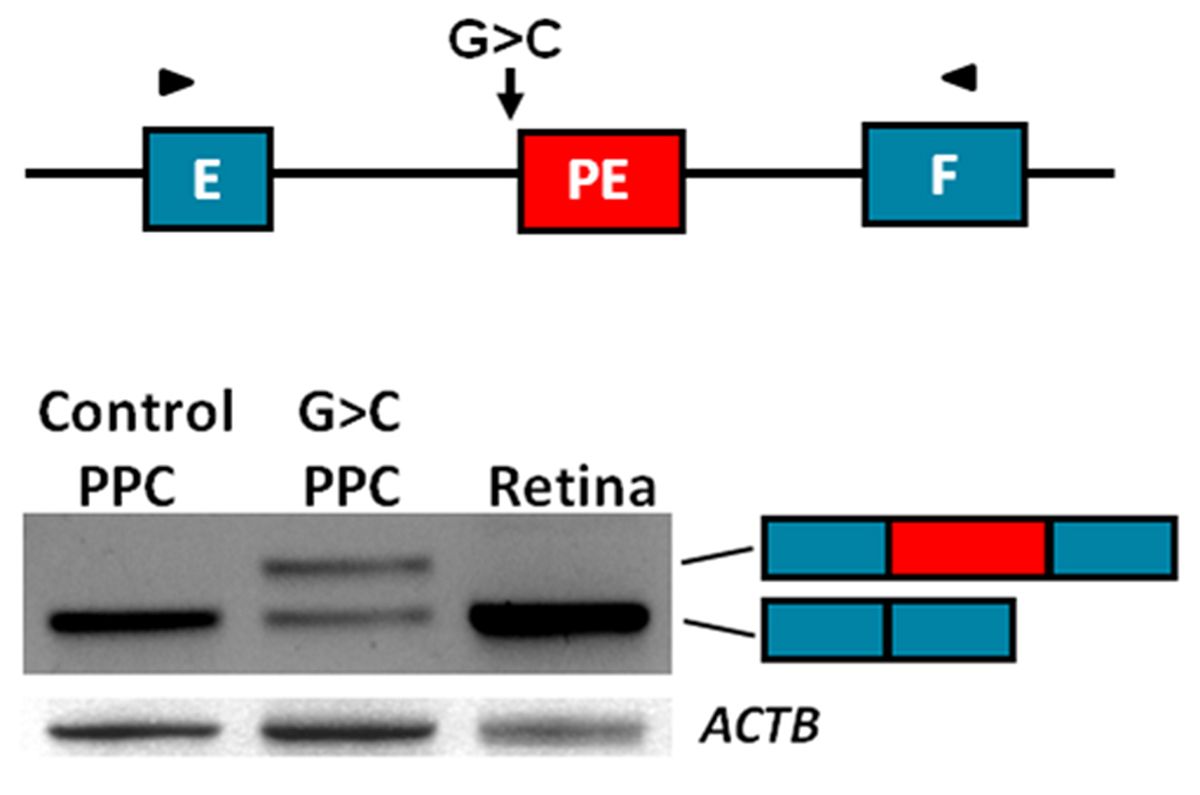
We generated iPSCs and PPCs for 15 cases with STGD1 or arCRD in which only a single causal variant was identified in the 50 exons of ABCA4. In parallel, in collaboration with prof. E. de Baere and M. Bauwens in Ghent, we sequenced the complete 128-kb ABCA4 gene in these and 65 other mono-allelic STGD1 and arCRD cases to reveal the mutation located in trans. We correlated the identified DNA variants with RNA defects using RT-PCR of mRNA extracted from patient-derived PPCs, and were able to identify several novel causal deep-intronic variants. Most of these activate or introduce novel splice sites and thereby generate pseudo-exons that are inserted into the ABCA4 mRNA (Riccardo Sangermano et al. unpublished data) (Figure 3).
 |
Designing a high-throughput cost-effective sequencing method
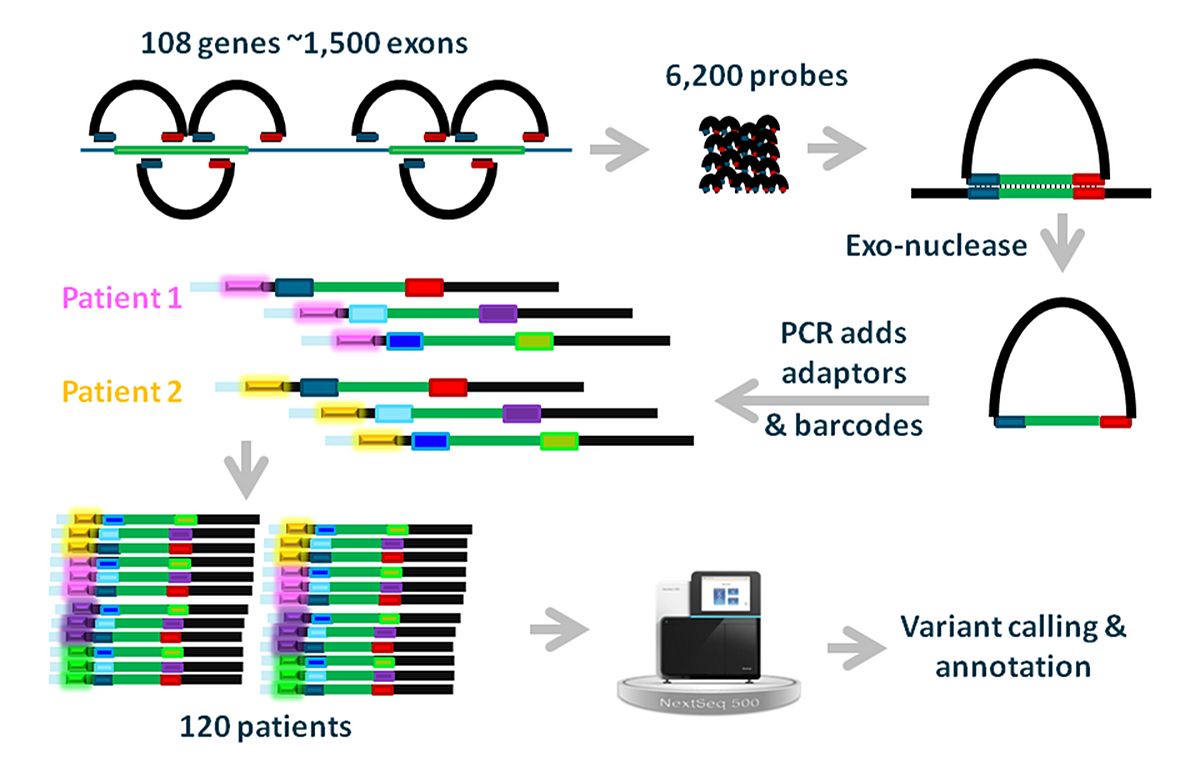
Although NGS-based sequencing costs are rapidly declining, targeted sequencing of coding regions of up to ~100 genes, or of coding and non-coding regions of single genes, is still relatively expensive. We therefore employed a cost-effective targeted sequencing technique based on molecular inversion probes (MIPs) to sequence the coding regions of 110 non-syndromic IRD-associated genes in more than 3.200 unsolved IRD cases from the European Retinal Disease Consortium (ERDC; www.erdc.info)(M. Imran Khan et al. unpublished data; Figure 4).
In this way we were able to solve up to 50% of cases who previously were prescreened for mutations in selected IRD genes and up to 75% of novel IRD probands. This targeted sequencing platform will continue to be operational for cheap (~ € 110,-/sample = € 1,-/gene/sample) research-based sequencing of IRD cases with RP, CRD, STGD1, Leber congenital amaurosis or achromatopsia.
 |
Future studies
Not only transcriptomics may enable us to interpret genomic variants. An unexplored area is metabolomics of blood plasma which may also provide hints towards the genetic defects or the functional pathways which are affected by the primary mutations.
Large scale genomics and transcriptomics studies may reveal more complex genetic interactions, such as in digenic inheritance,14 but also be able to address technically very challenging issues such as the role of genetic modifiers on expression and penetrance of IRDs.
Through the large scale sequencing of unsolved STGD1 cases, we aim to identify the majority of deep-intronic variants that result in pseudo-exons in ABCA4 mRNA. Through studies in the group of Dr. Rob Collin in Nijmegen, we will then will continue to design therapies to counteract the effect of these variants using antisense oligonucleotides that block their inclusion in the mRNA.
References (from 288 peer-reviewed papers)
- Cremers, F. P. M., van de Pol, T. J. R., van Kerkhoff, E. P. M., Wieringa, B. & Ropers, H.-H. Cloning of a gene that is rearranged in patients with choroideremia (1990) Nature 347, 674-677.
- Berger, W., van de Pol, T. J. R., Warburg, M., Gal, A., Bleeker-Wagemakers, E. M., de Silva, H., Meindl, A., Meitinger, T., Cremers, F. P. M. & Ropers, H.-H. Mutations in the candidate gene for Norrie disease (1992) Hum. Mol. Genet. 1, 461-465.
- Roepman, R., van Duynhoven, G., Rosenberg, T., Pinckers, A. J. L. G., Bleeker-Wagemakers, E. M., Bergen, A. A. B., Post, J., Beck, A., Reinhardt, R., Ropers, H.-H., Cremers, F. P. M. & Berger, W. Positional cloning of the gene for X-linked retinitis pigmentosa: homology with the guanine-nucleotide-exchange factor RCC1 (1996) Hum. Mol. Genet. 5, 1035-1041.
- Schwahn, U., Lenzner, S., Dong, J., Feil, S., Hinzmann, B., van Duijnhoven, G., Kirschner, R., Hemberger, M., Bergen, A. A. B., Rosenberg, T., Pinckers, A. J. L. G., Fundele, R., Rosenthal, A., Cremers, F. P. M., Ropers, H.-H. & Berger, W. Positional cloning of the gene for X-linked retinitis pigmentosa 2 (1998) Nature Genet. 19, 327-332.
- den Hollander, A. I., ten Brink, J. B., de Kok, Y. J. M., van Soest, S., van den Born, L. I., van Driel, M. A., van de Pol, T. J. R., Payne, A. M., Bhattacharya, S. S., Kellner, U., Hoyng, C. B., Westerveld, A., Brunner, H. G., Bleeker-Wagemakers, E. M., Deutman, A. F., Heckenlively, J. R., *Cremers, F. P. M. & Bergen, A. A. B. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12) (1999) Nature Genet. 23, 217-221. *Corresponding author.
- den Hollander, A. I., Koenekoop, R. K., Yzer, S., Lopez, I., Arends, M. L., Voesenek, K. E. J., Zonneveld, M. N., Strom, T. M., Meitinger, T., Brunner, H. G., Hoyng, C. B., van den Born, L. I., Rohrschneider, K. & Cremers, F. P. M. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. (2006) Am. J. Hum. Genet. 79, 556-561.
- den Hollander, A. I., Koenekoop, R. K., Mohamed, M. D., Arts, H. H., Boldt, K., Towns, K. V., Sedmak, T., Beer, M., Nagel-Wolfrum, K., McKibbin, M., Dharmaraj, S., Lopez, I., Ivings, L., Williams, G. A., Springell, K., Woods, C. G., Jafri, H., Rashid, Y., Strom, T. M., van der, Z. B., Gosens, I., Kersten, F. F., van, W. E., Veltman, J. A., Zonneveld, M. N., van Beersum, S. E., Maumenee, I. H., Wolfrum, U., Cheetham, M. E., Ueffing, M., *Cremers, F. P. M., *Inglehearn, C. F. & *Roepman, R. Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. (2007) Nat. Genet. 39, 889-895. (*equal contributions)
- Collin, R. W. J., Littink, K. W., Klevering, B. J., van den Born, L. I., Koenekoop, R. K., Zonneveld-Vrieling, M. N., Blokland, E. A. W., Strom, T. M., Hoyng, C. B., den Hollander, A. I. & Cremers, F. P. M. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. (2008) Am. J. Hum. Genet. 83, 594-603.
- *Roosing, S., *Lamers, I. J. C.., *de Vrieze, E., *van den Born, L. I., Lambertus, S., Arts, H. H., Study group [Boldt, K., de Baere, E., Klaver, C. C. W., Coppieters, F., Koolen, D. A., Lugtenberg, D., Neveling, K., van Reeuwijk, J., Ueffing, M., van Beersum, S. E. C., Zonneveld-Vrieling, M. N.], Peters, T. A., Hoyng, C. B., Kremer, H., Hetterschijt, L., Letteboer, S. J. F., #van Wijk, E., #Roepman, R., #den Hollander, A. I. & #Cremers, F. P. M. Disruption of the basal body protein POC1B results in autosomal recessive cone-rod dystrophy (2014). Am. J. Hum. Genet. 95, 131-142. *Shared first authors; #shared senior authors.
- Nikopoulos, K., Gilissen, C., Hoischen, A., van Nouhuys, C. E., Boonstra, F. N., Blokland, E. A. W., Arts, P., Wieskamp, N., Strom, T. M., Ayuso, C., Tilanus, M. A. D., Bouwhuis, S., Mukhopadhyay, A., Scheffer, H., Hoefsloot, E. H., Veltman, J. A., *Cremers, F. P. M. & Collin R. W. J. Next-generation sequencing of a 40 Mb linkage interval reveals TSPAN12 mutations in patients with familial exudative vitreoretinopathy. (2010) Am. J. Hum. Genet. 86, 240-247. *Corresponding author.
- Collin, R. W. J., Nikopoulos, K., Dona, M., Gilissen, C., Hoischen, A., Boonstra, F. N., Poulter, J. A., Kondo, H., Berger, W., Toomes, C., Tahira, T., Mohn, L. R., Blokland, E. A., Hetterschijt, L., Ali, M., Groothuismink, J. M., Duijkers, L. E. M, Inglehearn, C. F., Sollfrank, L., Strom, T. M., Uchio, E., van Nouhuys, C. E., Kremer, H., Veltman, J. A., van Wijk, E. & Cremers, F. P. M. ZNF408 is mutated in familial exudative vitreoretinopathy and crucial for the development of zebrafish retinal vasculature (2013). Proc. Natl. Acad. Sci. USA, 110, 9856-9861.
- Haer-Wigman, L., van Zelst-Stams, W. A., Pfundt, R., van den Born, L. I., Klaver, C. C., Verheij, J. B., Hoyng, C. B., Breuning, M. H., Boon, C. J., Kievit, A. J., Verhoeven, V. J., Pott, J. W., Sallevelt, S. C., van Hagen, J. M., Plomp, A. S., Kroes, H. Y., Lelieveld, S. H., Hehir-Kwa, J. Y., Castelein, S., Nelen, M., Scheffer, H., Lugtenberg, D., Cremers, F. P. M., Hoefsloot, L., Yntema, H. G. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. (2017) Eur J Hum Genet. 25:591-9.
- Sangermano, R., Bax, N. M., Bauwens, M., van den Born, L. I., De Baere, E., Garanto, A., Collin, R. W. J., Goercharn-Ramlal, A. S. A., den Engelsman-van Dijk, A. H. A., Rohrschneider, K., Hoyng, C. B., *Cremers, F. P. M., *Albert, S. Photoreceptor progenitor mRNA analysis reveals exon skipping due to the ABCA4 c.5461-10T>C mutation in Stargardt disease. (2016) Ophthalmology. 123:1375-85. *Shared senior authors.
- *Liu, Y. P., *Bosch, D. G. M., Siemiatkowska, A. M., Rendtorff, N. D., Boonstra, F. N., Möller, C., Tranebjærg, L., #Katsanis, N., #Cremers, F. P. M. Putative digenic inheritance of heterozygous RP1L1 and C2orf71 null mutations in syndromic retinal dystrophy. (2016) Ophthalmic Genet., 38:127-32. *Shared first authors; #shared senior authors.
Frans P.M. Cremers, PhD
Professor in Ophthalmogenetics
Department of Human Genetics & Donders Institute for Brain, Cognition and Behaviour
Radboudumc
Contact
Radboudumc
PO Box 9101, 6500 HB Nijmegen, The Netherlands
Phone: +31-24-3614017
E-mail: frans.cremers[at]radboudumc.nl
URL: http://www.ru.nl/donders/research/theme-2-perception/blindness-genetics/