vision-research.eu - The Gateway to European Vision Research
![]()

![]()
You are here: vision-research.eu » Vision Research » Visionary of the Quarter » Peter Humphries (Q01-2015)
The research topics of Peter Humphries
 |
Globally, over 160 million people are estimated to be visually impaired, and it is a lamentable fact that up to 70% of global visual handicap, caused by cataract, glaucoma, corneal opacities and infection, is largely treatable or preventable (including most open-‐angle glaucomas). In the developed World, conditions that are essentially eclipsed by the vastly more common forms of world blindness become very much more visible. These are the hereditary retinopathies (prominent among which is retinitis pigmentosa), age-‐related macular degeneration (AMD) and proliferative diabetic retinopathy. Also, up to 7% of cases of open-‐angle glaucoma either do not respond, or become resistant to conventional medications. For almost three decades, Pete’s research has been directed toward understanding the molecular pathologies associated with retinal degeneration, early studies focusing almost exclusively on hereditary retinopathies, the most common cause of registered visual handicap among those of working age in the developed world. On the back of a progressively developing human genetic linkage map, many groups began to focus attention on characterizing large families with hereditary retinopathies that could form the basis of gene localization studies.
An early break came in 1989, when, after about four years having been spent in RFLP screening of members of a large family with a dominant form of retinitis pigmentosa, a location for a dominant RP gene was established (McWilliam, P. et al, Genomics, 1989). Here is the abstract and sections of discussion from that paper: “Members of a large pedigree of Irish origin presenting with early onset Type I autosomal dominant retinitis pigmentosa (ADRP) have been typed for D3S47 (C17), a polymorphic marker from the long arm of chromosome 3. Significant, tight linkage of ADRP to D3S47, with a lod score of 14.7 maximizing at 0.00 recombination, has been obtained, hence localizing the ADRP gene (RP1) segregating in this pedigree to 3q…… rhodopsin, the major integral membrane protein of the rod outer segments, has been assigned to 3q21-‐3q24….it is therefore interesting to speculate that rhodopsin and/or retinol binding protein 1 or 2 may be potential candidate genes for ADRP”.
Subsequently, other locations began to emerge in similar studies of large dominant RP families from Ireland, including, for example, 6p and 7q, where mutations within genes encoding RDS-‐peripherin and inosine monophosphate dehydrogenase were identified (reviewed Humphries et al, Science, 1992). As a result of world efforts, it is now clear that hereditary retinopathies possibly represent the most genetically heterogeneous of any group of hereditary conditions for which molecular pathologies have been explored. On the one hand, the progressive unfolding of an immensely complex genetic landscape has been truly inspirational, but on the other, and in a very much more translational sense, this same genetic heterogeneity presents an immensely significant challenge – developing gene medicines for what could be several hundred individually rather rare hereditary conditions is a major logistical and economic hurdle that must now be surmounted.
Indeed a major recent focus of research has been in the development of procedures for modulating permeability at the inner blood retina barrier such as to allow access to the retina of systemically administered low molecular weight potentially protective compounds targeting molecular pathologies common to multiple forms of disease (protein misfolding/aggregation; oxidative stress, neuroprotection etc) which could be used either singly or in a combinatorial sense together with gene therapies (see figure 1 and Campbell et al, PNAS 2009; Campbell et al, EMBO Molec Med 2011; Campbell et al., Nature Communications, 2012).
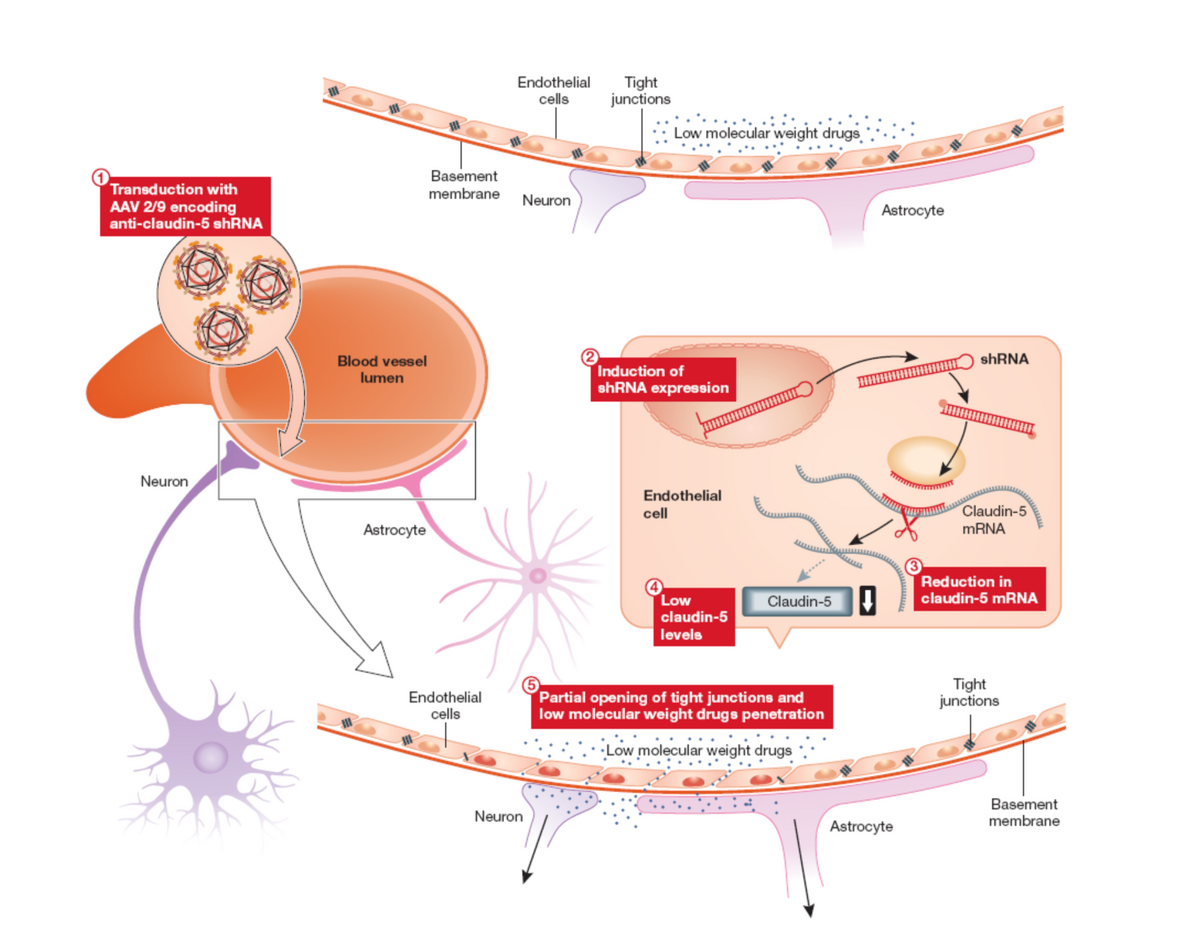 |
More recently, multifactorial conditions, including AMD and glaucoma have become a major focus of research. Genetic risk factors associated with AMD in genes encoding (among many others) components of the complement cascades are well known. Drusen is present in all cases of AMD and it has now been shown that the NLRP3 inflammasome plays a protective role in AMD through the induction of the cytokine, IL-‐18 by drusen components. (see Figure 2 and Doyle et al., Nature Medicine 2012). Therapies based on IL-‐18-‐mediated suppression of choroidal neovascularisation are now being acitvely explored (Campbell et al, Science Translational Medicine, 2014).
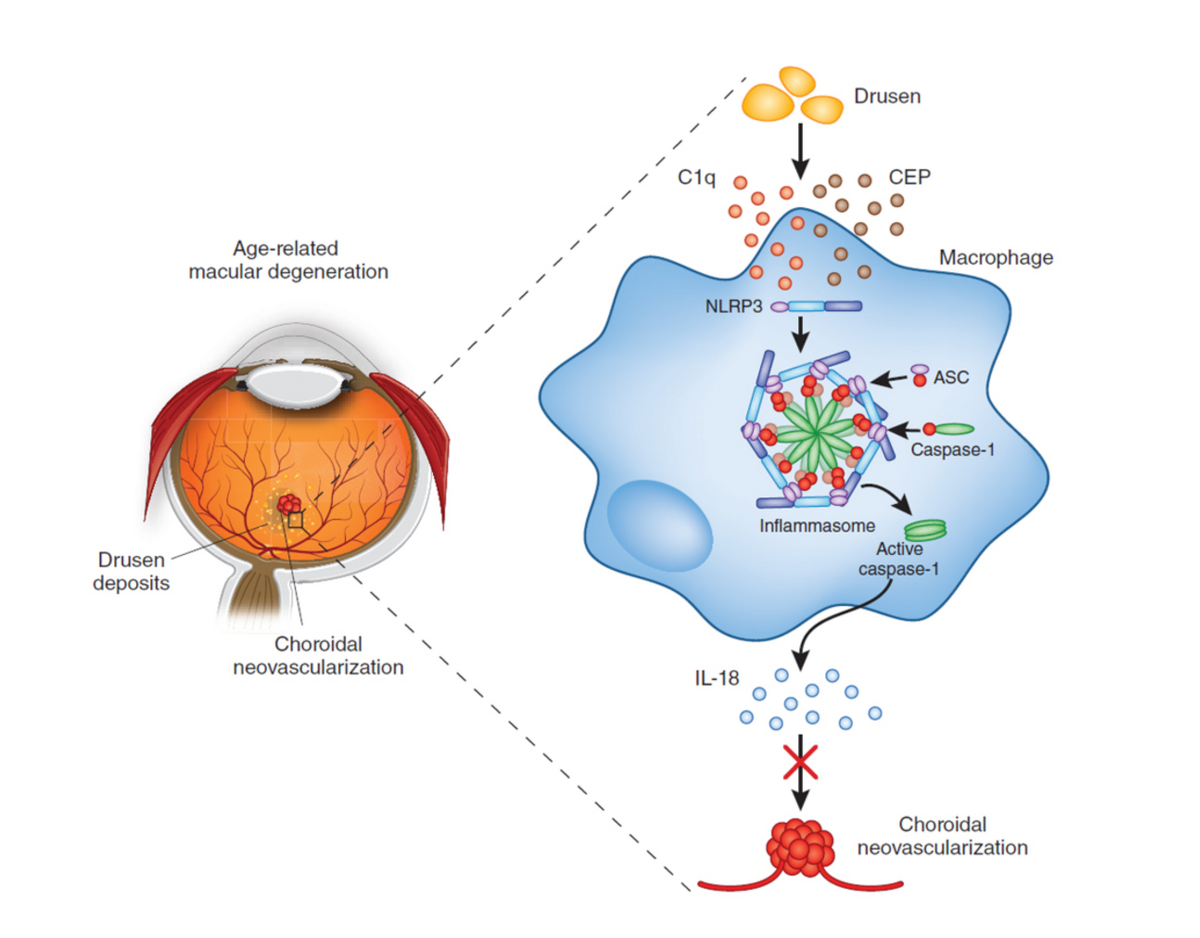 |
Prof. Peter Humphries
- Professor of Medical Molecular Genetics
- vision-research.eu-Profile
Contact
University of Dublin
Trinity College Dublin
Lincoln Place Gate
Dublin 2
D2 Dublin, Ireland
Phone: +353-1-8961547
Fax: +353-1-8963848
Email: pete.humphries[at]tcd.ie
Website: www.tcd.ie