vision-research.eu - The Gateway to European Vision Research
![]()

![]()
You are here: vision-research.eu » Vision Research » Visionary of the Quarter » Elfride De Baere (Q01-2020)
Precision medicine to understand missing heritability in inherited retinal diseases
 |
The major topics of the research work of Professor Elfride De Baere
Inherited retinal diseases (IRD) are a major cause of early-onset blindness, affecting over two million people worldwide. They are characterized by a tremendous clinical and genetic heterogeneity, with over 270 disease genes identified (https://sph.uth.edu/retnet).
The major topics of my research deal with precision medicine in IRD (Figure 1).
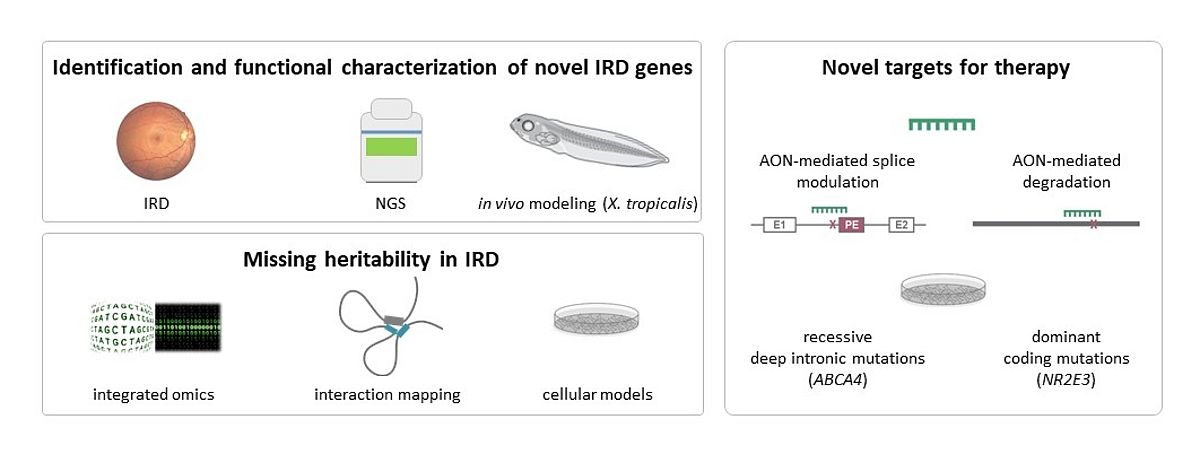 |
Identification and functional characterization of novel IRD genes
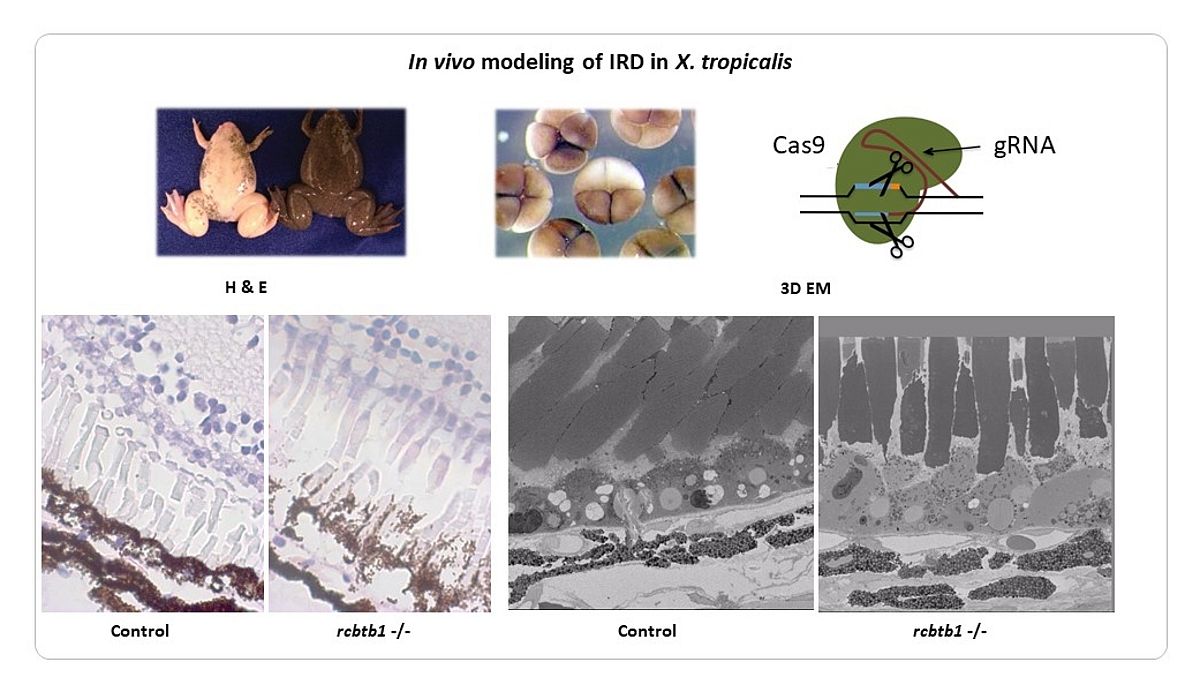
We have identified novel genes implicated in IRD using a variety of genomic strategies (linkage studies and Sanger sequencing, candidate gene testing, homozygosity mapping, whole exome sequencing [WES]). Some of these genes play a role in general processes such as transcription (NR2E3, RAX2) (1,2), splicing (SF3B2) (Van Cauwenbergh, Van de Sompele et al. in preparation) and ubiquitination (RCBTB1) (3). The identification of additional families has been facilitated by the European Retinal Disease Consortium (ERDC) (www.erdc.info). To shed light on the molecular pathogenesis we have generated knockdown (sf3b2) (Van Cauwenbergh, Van de Sompele et al. in preparation) and knockout (rcbtb1, abca4, ush2a) models in Xenopus tropicalis (Figure 2) (Carron, Naert, Ascari et al. in preparation). The latter is a suitable model system for IRD, having the major cell types of the human eye and other characteristics that make it amenable for genome editing with high efficiency.
 |
Missing heritabilty in IRD
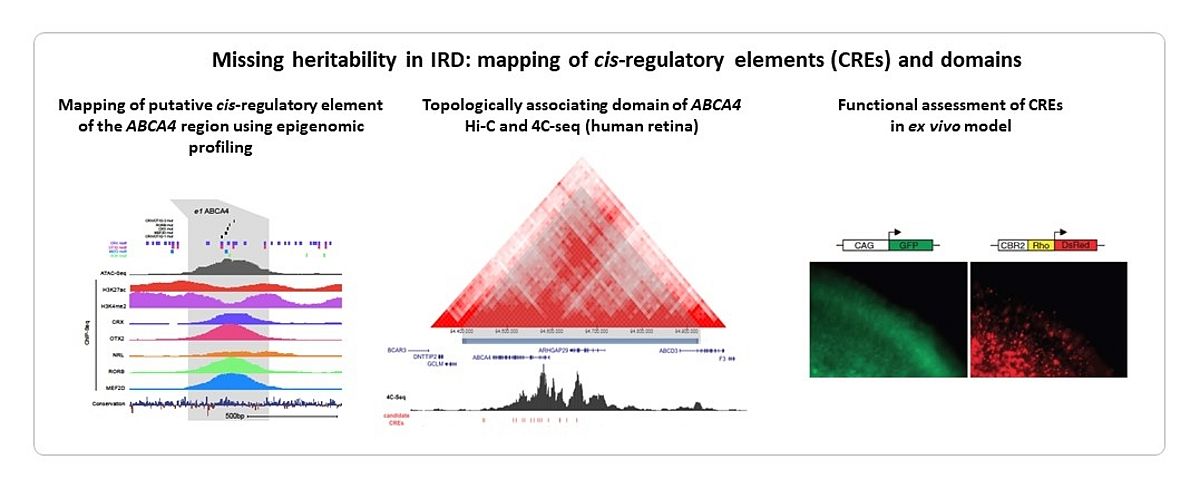
Despite large-scale testing of known IRD genes, ~50% of tested cases remain unsolved. We and others hypothesize that a large part of the missing heritability resides in the non-coding part of the genome that is not covered by exomic approaches. We provided proof-of-concepts for missing heritability in IRD. Copy number variants (CNVs) of IRD genes represent 5-10% of the mutational spectrum. We explored the genomic landscape of IRD genes to prioritize those genes susceptible to CNV formation (4). We used a customized microarray, arrEYE, for high-resolution CNV analysis of known and candidate IRD genes and of retina-expressed noncoding RNAs (ncRNAs) (5). The concept of non-coding variation in IRD has been strengthened by autosomal recessive monogenic IRD in which only a single heterozygous mutation can be found in the coding region of the expected disease-causing gene, the so-called monoallelic cases. We showed that a deep-intronic splice mutation in CEP290 accounts for ~20% of congenital blindness in Belgium (6). We identified missing variation in monoallelic patients with ABCA4-associated disease using targeted testing and ABCA4 locus resequencing (7,8,9). Deep-intronic splice variants including a Belgian founder, a recurrent milder variant, CNVs and variants with a putative effect on regulation were identified, revealing a molecular diagnosis in 84% of the cohort. IRD-associated regulatory variants affecting transcription are less studied. We linked 5’ untranslated region (5’UTR) regulatory mutations in NMNAT1 with Leber Congenital Amaurosis for the first time (10). As the interpretation of cis-regulatory variants is challenging, integrated cis-regulatory maps based on epigenome and transcriptome profiling in human retinal tissue and cells are required to close the gap between non-coding variants and IRD pathogenesis (11,12) (Figure 3).
 |
Novel therapeutic targets
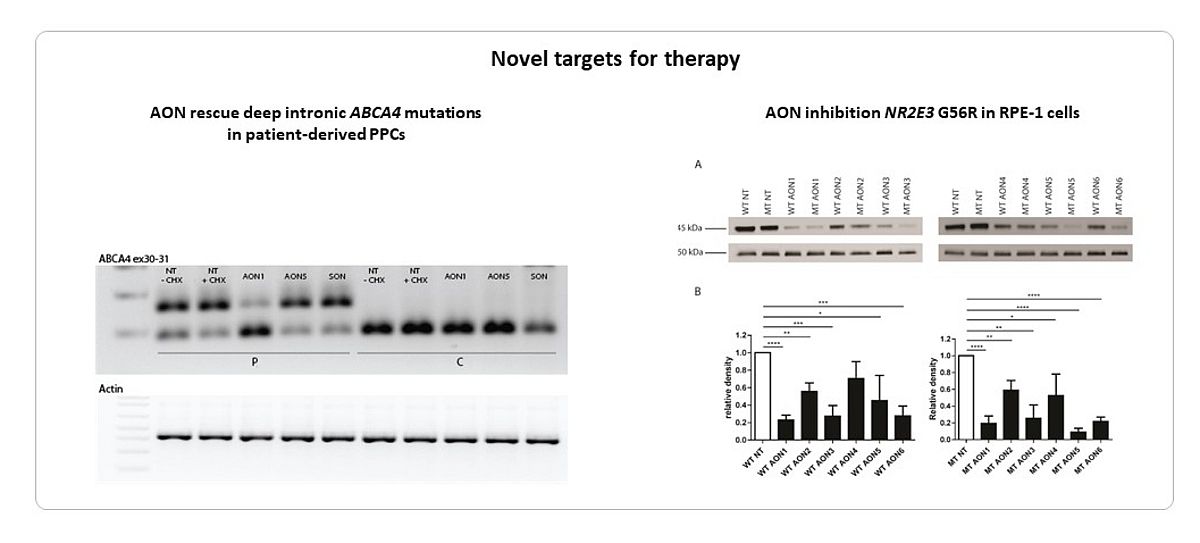
We were able to rescue the effect seven deep-intronic ABCA4 variants using antisense oligonucleotides (AONs) (8,9). We showed that two adjacent mutations, c.4539+1100A>G and c.4539+1106C>T, could be corrected by the same AON using patient-derived photoreceptor precursor cells (PPCs) (Naessens et al. in preparation). We targeted a recurrent, dominant negative allele (c.166G>A) in NR2E3 using AONs, aiming at a specific downregulation of the mutated allele. We identified a subtle, mutant-specific downregulation for AONs containing specialized modifications (13). Further research is ongoing to improve AON specificity however (Figure 4).
 |
References
- Coppieters F, Leroy BP, Beysen D, Hellemans J, De Bosscher K, Haegeman G, Robberecht K, Wuyts W, Coucke PJ, De Baere E. Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa. Am J Hum Genet. 2007 Jul;81(1):147-57. Epub 2007 May 24. PubMed PMID: 17564971; PubMed Central PMCID: PMC1950922.
- Van de Sompele S, Smith C, Karali M, Corton M, Van Schil K, Peelman F, Cherry T, Rosseel T, Verdin H, Derolez J, Van Laethem T, Khan KN, McKibbin M, Toomes C, Ali M, Torella A, Testa F, Jimenez B, Simonelli F, De Zaeytijd J, Van den Ende J, Leroy BP, Coppieters F, Ayuso C, Inglehearn CF, Banfi S, De Baere E. Biallelic sequence and structural variants in RAX2 are a novel cause for autosomal recessive inherited retinal disease. Genet Med. 2018 Oct 31. doi: 10.1038/s41436-018-0345-5. PubMed PMID: 30377383.
- Coppieters F, Ascari G, Dannhausen K, Nikopoulos K, Peelman F, Karlstetter M, Xu M, Brachet C, Meunier I, Tsilimbaris MK, Tsika C, Blazaki SV, Vergult S, Farinelli P, Van Laethem T, Bauwens M, De Bruyne M, Chen R, Langmann T, Sui R, Meire F, Rivolta C, Hamel CP, Leroy BP, De Baere E. Isolated and Syndromic Retinal Dystrophy Caused by Biallelic Mutations in RCBTB1, a Gene Implicated in Ubiquitination. Am J Hum Genet. 2016 Aug 4;99(2):470-80. doi: 10.1016/j.ajhg.2016.06.017. PubMed PMID: 27486781; PubMed Central PMCID: PMC4974088.
- Van Schil K, Naessens S, Van de Sompele S, Carron M, Aslanidis A, Van Cauwenbergh C, Kathrin Mayer A, Van Heetvelde M, Bauwens M, Verdin H, Coppieters F, Greenberg ME, Yang MG, Karlstetter M, Langmann T, De Preter K, Kohl S, Cherry TJ, Leroy BP; CNV Study Group, De Baere E. Mapping the genomic landscape of inherited retinal disease genes prioritizes genes prone to coding and noncoding copy-number variations. Genet Med. 2018 Feb;20(2):202-213. doi: 10.1038/gim.2017.97. Epub 2017 Jul 27. PubMed PMID: 28749477; PubMed Central PMCID: PMC5787040.
- Van Cauwenbergh C, Van Schil K, Cannoodt R, Bauwens M, Van Laethem T, De Jaegere S, Steyaert W, Sante T, Menten B, Leroy BP, Coppieters F, De Baere E. arrEYE: a customized platform for high-resolution copy number analysis of coding and noncoding regions of known and candidate retinal dystrophy genes and retinal noncoding RNAs. Genet Med. 2017 Apr;19(4):457-466. doi: 10.1038/gim.2016.119. Epub 2016 Sep 8. PubMed PMID: 27608171; PubMed Central PMCID: PMC5392597.
- Coppieters F, Casteels I, Meire F, De Jaegere S, Hooghe S, van Regemorter N, Van Esch H, Matuleviciene A, Nunes L, Meersschaut V, Walraedt S, Standaert L, Coucke P, Hoeben H, Kroes HY, Vande Walle J, de Ravel T, Leroy BP, De Baere E. Genetic screening of LCA in Belgium: predominance of CEP290 and identification of potential modifier alleles in AHI1 of CEP290-related phenotypes. Hum Mutat. 2010 Oct;31(10):E1709-66. doi: 10.1002/humu.21336. PubMed PMID: 20683928; PubMed Central PMCID: PMC3048164.
- Bauwens M, De Zaeytijd J, Weisschuh N, Kohl S, Meire F, Dahan K, Depasse F, De Jaegere S, De Ravel T, De Rademaeker M, Loeys B, Coppieters F, Leroy BP, De Baere E. An augmented ABCA4 screen targeting noncoding regions reveals a deep intronic founder variant in Belgian Stargardt patients. Hum Mutat. 2015 Jan;36(1):39-42. doi: 10.1002/humu.22716. PubMed PMID: 25346251.
- Bauwens M, Garanto A, Sangermano R, Naessens S, Weisschuh N, De Zaeytijd J, Khan M, Sadler F, Balikova I, Van Cauwenbergh C, Rosseel T, Bauwens J, De Leeneer K, De Jaegere S, Van Laethem T, De Vries M, Carss K, Arno G, Fakin A, Webster AR, de Ravel de l'Argentière TJL, Sznajer Y, Vuylsteke M, Kohl S, Wissinger B, Cherry T, Collin RWJ, Cremers FPM, Leroy BP, De Baere E. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet Med. 2019 Jan 23. doi: 10.1038/s41436-018-0420-y. PubMed PMID: 30670881.
- Sangermano R, Garanto A, Khan M, Runhart EH, Bauwens M, Bax NM, van den Born LI, Khan MI, Cornelis SS, Verheij JBGM, Pott JR, Thiadens AAHJ, Klaver CCW, Puech B, Meunier I, Naessens S, Arno G, Fakin A, Carss KJ, Raymond FL, Webster AR, Dhaenens CM, Stöhr H, Grassmann F, Weber BHF, Hoyng CB, De Baere E, Albert S, Collin RWJ, Cremers FPM. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet Med. 2019 Jan 15. doi: 10.1038/s41436-018-0414-9. PubMed PMID: 30643219.
- Coppieters F, Todeschini AL, Fujimaki T, Baert A, De Bruyne M, Van Cauwenbergh C, Verdin H, Bauwens M, Ongenaert M, Kondo M, Meire F, Murakami A, Veitia RA, Leroy BP, De Baere E. Hidden Genetic Variation in LCA9-Associated Congenital Blindness Explained by 5'UTR Mutations and Copy-Number Variations of NMNAT1. Hum Mutat. 2015 Dec;36(12):1188-96. doi: 10.1002/humu.22899. Epub 2015 Oct 1. PubMed PMID: 26316326; PubMed Central PMCID: PMC5054839.
- Van Schil K, Karlstetter M, Aslanidis A, Dannhausen K, Azam M, Qamar R, Leroy BP, Depasse F, Langmann T, De Baere E. Autosomal recessive retinitis pigmentosa with homozygous rhodopsin mutation E150K and non-coding cis-regulatory variants in CRX-binding regions of SAMD7. Sci Rep. 2016 Feb 18;6:21307. doi: 10.1038/srep21307. PMID: 26887858.
- Cherry TJ, Yang MG, Harmin DA, Tao P, Timms AE, Bauwens M, Allikmets R, Jones EM, Chen R, De Baere E, Greenberg ME. bioRxiv doi: doi.org/10.1101/412361
- Naessens S, Ruysschaert L, Lefever S, Coppieters F, De Baere E. Antisense Oligonucleotide-Based Downregulation of the G56R Pathogenic Variant Causing NR2E3-Associated Autosomal Dominant Retinitis Pigmentosa. Genes. 2019 May 10;10(5). pii: E363. doi: 10.3390/genes10050363. PMID: 31083481.
 |
Professor Elfride De Baere
ORCID: 0000-0002-5609-6895
Center for Medical Genetics Ghent (CMGG)
Campus Ghent University Hospital, MRB1,
Corneel Heymanslaan 10
9000 Ghent, Belgium
Email: Elfride.DeBaere[at]UGent.be
Website: www.debaerelab.com | www.startn.eu
Twitter: @elfridedebaere