vision-research.eu - The Gateway to European Vision Research
![]()

![]()
You are here: vision-research.eu » Vision Research » Visionary of the Quarter » Enrica Strettoi (Q02-2020)
It's all in the eyes: basic science approaches to understand and treat inherited retinal degeneration
 |
The research work of Professor Dr. Enrica Strettoi
It has been said that the eye is such a marvelous organ that microscopes and telescopes only expand its authority. Indeed, the fascinating aspect of laboratory work for me begins with the esthetic pleasure I feel when I look at the retina through a microscope: a dissecting binocular, where the retina appears thin and transparent like a butterfly wing, or the amazing laser confocal, where images show colorful neuronal processes entangled in synaptic connections, to end with the unsurpassed elegance and resolution of the electron microscope, where cells, organelles and junctions show their amazing details. Since my student years, my research starts with microscopical observations of the retina, and then combines morphology with image analysis, in vivo and ex vivo pharmacological treatments, molecular biology, and behavioral tools with the goal of understanding and treating retinal neurodegenerative diseases.
The normal retina begins performing its functions by splitting light signals in parallel channels carrying to the visual part of the brain information pertinent to brightness, contrast, color, motion, differences in time domain etc.; yet, this perfect structural organization and computational ability, endowed in about 60 different neuronal types confined in a space as thin as a quarter of a millimeter, can be lost when photoreceptors die out, as in the case of Retinitis Pigmentosa (RP). This is a family of genetic disorders caused by hundreds different mutations in heterogeneous genes, coding for very different proteins, which encompass phototransduction enzymes, visual cycle proteins, structural constituents of cilia as well as transcription factors and others cellular components and regulators. In RP, usually rods carry the mutation and die first, so that night blindness develops. Later (and for still partially unknown reasons) cones also die out. It is because of this “bystander effect” that diurnal, high acuity vision is lost in RP, a blinding disease with yet limited possibilities of cure and at the same time biologically intriguing: why do cells that do not express the mutation (i.e. the cones) die out? And what happens in the retina “beyond” photoreceptors? How does the exquisite architecture of inner retinal neurons react to the death of the main input cells (rods and cones), and how adverse remodeling of inner retinal circuitry affect our ability to restore vision?
Environment and inflammation
Studies carried on in my laboratory in the last years (Barone et al., 2012, 2014; Guadagni et al., 2015) have shown the environment can modulate RP progression, exactly as it does in other neurodegenerative disease, typically Alzheimer and Huntington. Environmental Enrichment (EE) is an experimental protocol by which laboratory animals (typically rodents) are exposed to an enhanced sensory and social stimulation and increased physical activity. This no-harm manipulation has powerful structural and functional effects, mostly studied at the hippocampal level, where neurogenesis, learning and memory are potentiated. In neurological conditions, an enriched environment can delay the aggressiveness and speed of progression of the disease. Because the retina is simply an outpost of the brain, we reasoned that it should react to EE like any other part of the CNS and when we enriched from birth rd10 mice, carrying a retinal degeneration mutation, we found that, indeed, the phenotype became milder and slower and the time window of useful vision correspondingly expanded.
These were the first studies to demonstrate a therapeutic action of EE for the degenerating retina and opened the path for subsequent rescue approaches based on physical exercise, one of the main components of EE. In the past, light has been the main environmental factor studied for its effects on RP progression; EE studies go definitely off the path and offer a non-invasive way to preserve a viable retina in view of a more definitive therapeutic approach.
In the search of molecular effectors of the protective retinal response triggered by EE, we took a transcriptome approach of the retina of normal, degenerating and enriched-exposed mice. We made an interesting discovery: in molecular terms, inflammation and immune responses constitute the prevalent biological processes in the retina at the peaks of rod and cone death. Local activation of microglia and macrophage recruitment contribute to this response, which has the effect of literally bathing the outer retina with cytokines and chemokines that alter the local milieu. We and others believe that this alteration contributes to disease worsening, particularly the secondary death of cones.
Noticeably, exposure to EE across the period of maximum photoreceptor death has the ability to lower signs of retinal inflammation, promote cone survival and delay vision loss. This effect can be mimicked by general administration of Dexamethasone, a well-known steroid with an inhibitory action on the immune response, already used in RP patients to treat macular edema, a common complication of the main disease. Studies with additional anti-inflammatory agents are presently ongoing in my lab: our effort is transforming this experimental works in clinical trials for RP patients, pursuing the goal of slowing down the inevitable loss of cone cells and visual acuity. Slowing down an intrinsically slow disease, such as RP, represents itself a cure and can improve the quality of life of many people, independently from the underlying mutations.
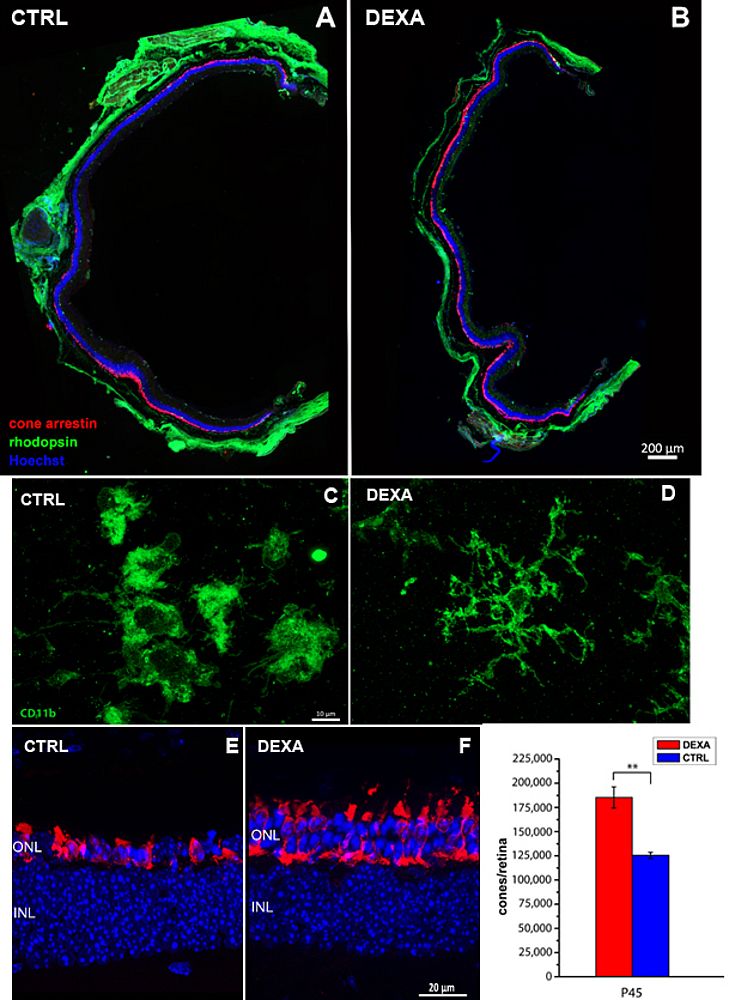 |
Retinal remodeling
A long-term line of investigation of my laboratory has been the study of remodeling in retinal degeneration, that is to say the search for secondary changes among neurons, glia and blood vessels that follow the primary process of photoreceptor death. Viability of the inner retina is crucial for successful implementation of implant of electronic prostheses and optogenetics vision restoration.
Disease remodeling is usually a regressive chain of events, starting subtly with retractions of dendrites and axons in bipolar cells and slowly leading to progressive atrophy and cell death in the inner retina, accompanied by formation of glial scars and blood vessel attenuation. But before these (almost unrepairable) changes, the inner retina shows some reactivity reminiscent of the plastic reaction of the injured spinal cord, where sprouting, migration and active formation of new synapses occur. Early remodeling is then an exploitable process to support regeneration or, at least, successful implantation of stem cells and a process favoring neuronal survival.
We recently studied remodeling in Tvrm4 mice, isolated at Jacksons laboratories, bearing a silent mutation on the 7th transmembrane domain of rhodopsin, which can be activated in adulthood upon a short exposure to bright light, mimicking time of onset of typical human RP.
Combining a panel of quantitative morphological and ultrastructural techniques, we demonstrated a high rate of bipolar cell survival despite regressive dendritic and axonal remodeling, considerable preservation of higher order neurons and moderate change in the number of ribbon synapses in the inner retina of adult Tvrm4 mice. Noticeably, dopaminergic amacrine cells sprout profusely in the outer retina, perhaps contributing to the genesis of spontaneous electrical activity reported in other retinal degeneration mutants. Altogether, secondary abnormalities found in Tvrm4 mutants during the immediate stage following photoreceptor loss coincide with previously described characteristics of other RP rodent models. Hence, we conclude that these remodeling features are independent of the age of disease onset and the underlying genetic mutation and are true hallmarks of RP.
The inducible phenotype observed in Tvrm4 mice mimics faithfully type B1 human RP with focal photoreceptor senescence and moderate speed of disease progression. The finding that remodeling in adult Tvrm4 mice is by no means more severe than in developmental models of the disease provides encouraging support for implementing retinal repair strategies for human RP, particularly optogenetics based on bipolar ells, which show complete survival in this mutant. We hope that the data we provided about remodeling will help to devise successful vision restoration approaches in the near future.
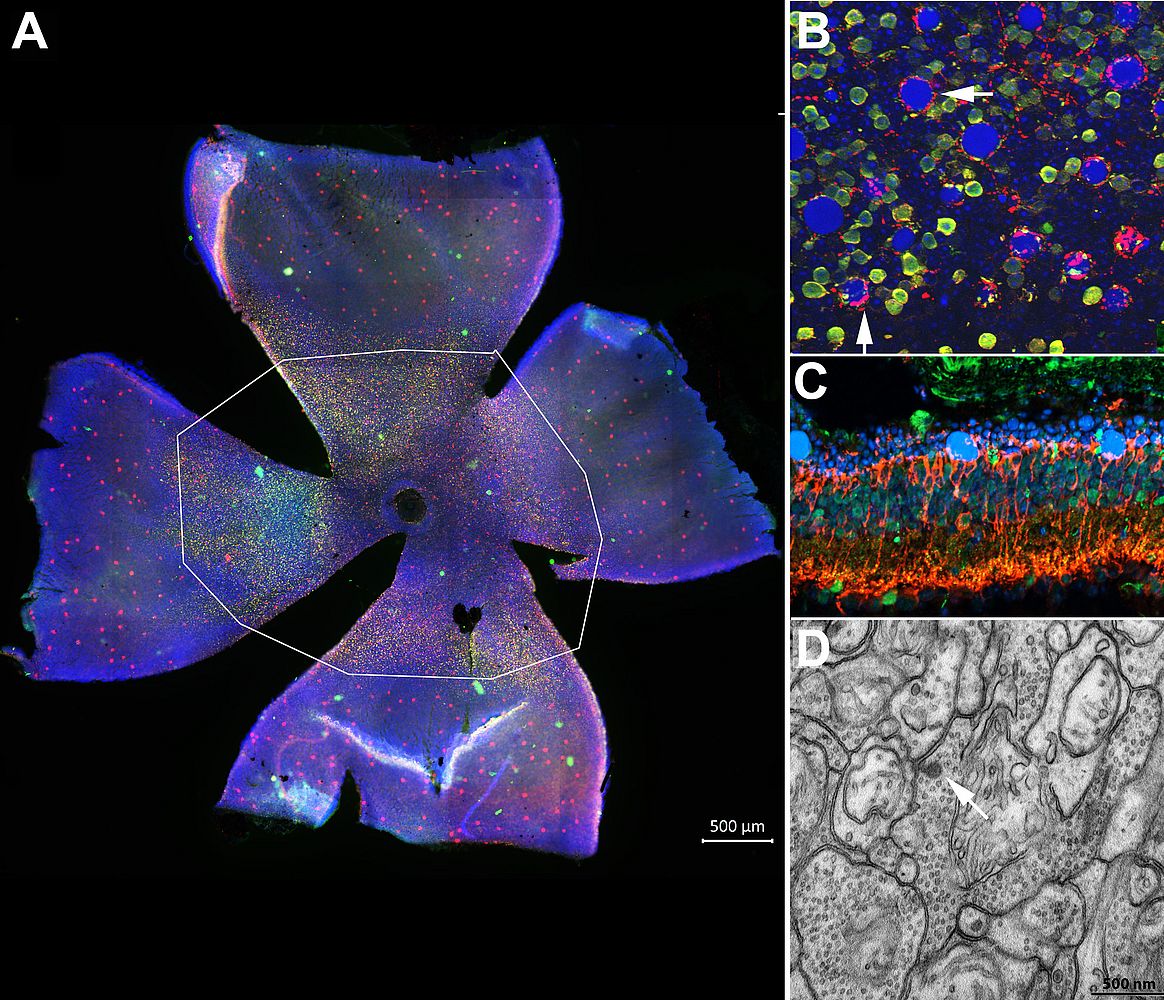 |
Representative publications
- Stefanov A, Novelli E, Strettoi E. Inner retinal preservation in the photoinducible I307N rhodopsin mutant mouse, a model of autosomal dominant retinitis pigmentosa. J Comp Neurol. 2019 Dec 6.
- Guadagni V, Biagioni M, Novelli E, Aretini P, Mazzanti CM, Strettoi E. Rescuing cones and daylight vision in retinitis pigmentosa mice. FASEB J. 2019 Sep;33(9):10177-10192.
- Strettoi E, Masri RA, Grünert U. AII amacrine cells in the primate fovea contribute to photopic vision. Sci Rep. 2018 Nov 6;8(1):16429.
- Gargini C, Novelli E, Piano I, Biagioni M, Strettoi E. Pattern of retinal morphological and functional decay in a light-inducible, rhodopsin mutant mouse. Sci Rep. 2017 Jul 18;7(1):5730.
- Guadagni V, Novelli E, Piano I, Gargini C, Strettoi E. Pharmacological approaches to retinitis pigmentosa: A laboratory perspective. Prog Retin Eye Res 2015 Sep;48:62-81.
- Barone I, Novelli E, Piano I, Gargini C, Strettoi E. Environmental enrichment extends photoreceptor survival and visual function in a mouse model of retinitis pigmentosa. PLoS One. 2012;7(11):e50726.
- Strettoi E, Gargini C, Novelli E, Sala G, Piano I, Gasco P, Ghidoni R. Inhibition of ceramide biosynthesis preserves photoreceptor structure and function in a mouse model of retinitis pigmentosa. Proc Natl Acad Sci U S A. 2010 Oct 26;107(43):18706-11.
- Mazzoni F, Novelli E, Strettoi E. Retinal ganglion cells survive and maintain normal dendritic morphology in a mouse model of inherited photoreceptor degeneration. J Neurosci. 2008 Dec 24;28(52):14282-92.
The lab and the collaborations
 |
I am a Director of Research at the CNR Neuroscience Institute in Pisa and Professor of Neurogenomics for Neuroscience Degree at the University of Pisa. My research is presently supported by the Velux Foundation (CH) and the EU.
I collaborate with Prof. Claudia Gargini, at the University of Pisa; with Prof. Antonino Cattaneo and Prof. Simona Capsoni, at the Scuola Normale Superiore of Pisa, with Prof. Riccardo Ghidoni, at the San Paolo Medical School of the University of Milan, and with Prof. Stanislao Rizzo and Prof. Benedetto Falsini at the Department of Ophthalmology of the Catholic University of Rome.

Professor Doctor Enrica Strettoi
Director of Research, Neuroscience Institute, Italian National Research Council (CNR), Pisa
Adjunct Professor of Neurogenomics, University of Pisa (UniPi)
Award Committee, Association for Research in Vision and Ophthalmology (ARVO), Member
CNR Neuroscience Institute
Area della Ricerca
Va Giuseppe Moruzzi 1
56124 Pisa, Italy
Tel/: +39 050 3153213
Email: enrica.strettoi[at]in.cnr.it
Website: http://www.in.cnr.it/index.php/it/people-it/150-enrica-strettoi