vision-research.eu - The Gateway to European Vision Research
![]()

![]()
You are here: vision-research.eu » Vision Research » Visionary of the Quarter » Hélène Dollfus (Q01-2010)
Challenges of inherited eye diseases

From the slit lamp side, it remains a terrible task to announce to a patient and his family that they will suffer from severe visual impairment for the rest of their life and that there is usually no radical treatments immediately accessible (although very recent progresses have emerged with gene therapy). Inherited eye disorders cover an extremely large spectrum of conditions ranging from abnormal eye development to progressive degenerative diseases. All of these have usually major consequences for the patients not only on the visual side but also on the inheritance side for the whole family. Identifying the genetic causes of this wide range of disorders remains a burden and is very important not only for genetic counselling purposes but also for understanding the underlying biological causes in order to understand the clinical presentation(s) and also as a first step to therapeutic approaches. Our centre for rare inherited disorders of the eye, is constantly aiming to the best genetic understanding and clinical care of this vast group of disorders (illustrated by overview 1).
From the bench side, studying inherited eye disorders irradiates on many biological pathways and makes this area of research extremely rich and challenging. Our group’s laboratory is focusing on the study of two types of inherited syndromic retinal dystrophies linked to two very different biological pathways: Bardet-Biedl syndrome (illustrated by overview 2), an emblematic ciliopathy and Cockayne syndrome due to transcription-repair deficiencies. Understanding the cause(s) of the retinal degeneration is these two examples gives good models for the mechanisms that lead to the death of the photoreceptor cells and will hopefully indicate targets for treating patients not only limited to the rare disorder group.
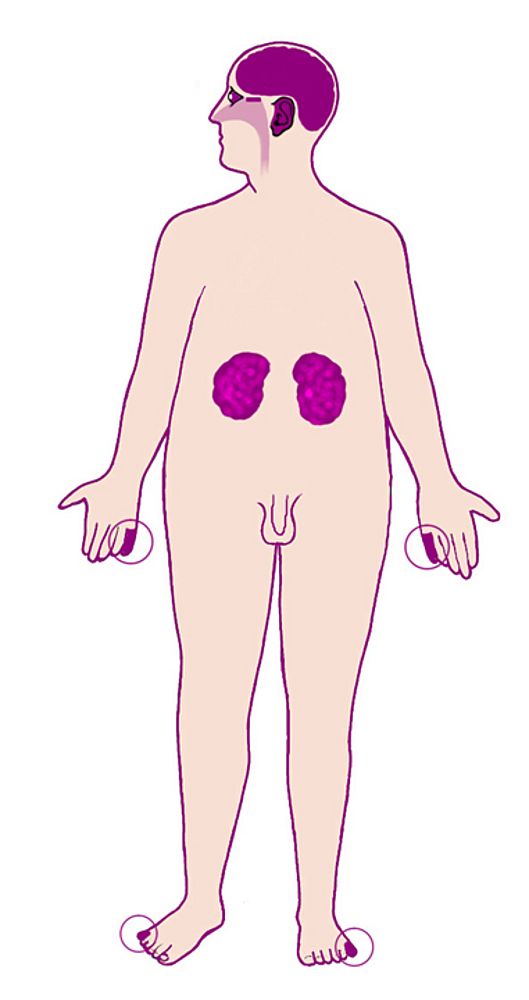
*Overview 1: Clinical and phenotype-genotype research
Clinical practice in the field of rare disorders often reaches clinical research as exceptional observations are frequently encountered and enables the constitution of interesting cohorts or cases. Although access to molecular testing is rapidly developing for routine molecular investigations such as for retinitis pigmentosa, phenotype-genotype correlations remain an important topic. Moreover, for example new approaches such as the use of micro arrays for chromosomal rearrangement detection widens our strategies. For instance, our group has recently studied a group of patients presenting with a very rare syndromic spectrum of facial and eye malformations (BOFS: Blepharo Oculo Facial Syndrome) and confirmed the implication of a transcription factor gene previously identified (TFAP2A). Studying phenotypes related to ciliopathies, presenting with retinitis pigmentosa, is another rapidely moving and fascinating field for which natural history studies are warranted. A vast majority of studies for rare eye conditions require enriching national and international collaborative studies which we have the pleasure to initiate or to contribute to by clinical research on relevant patients.
Figures
 |
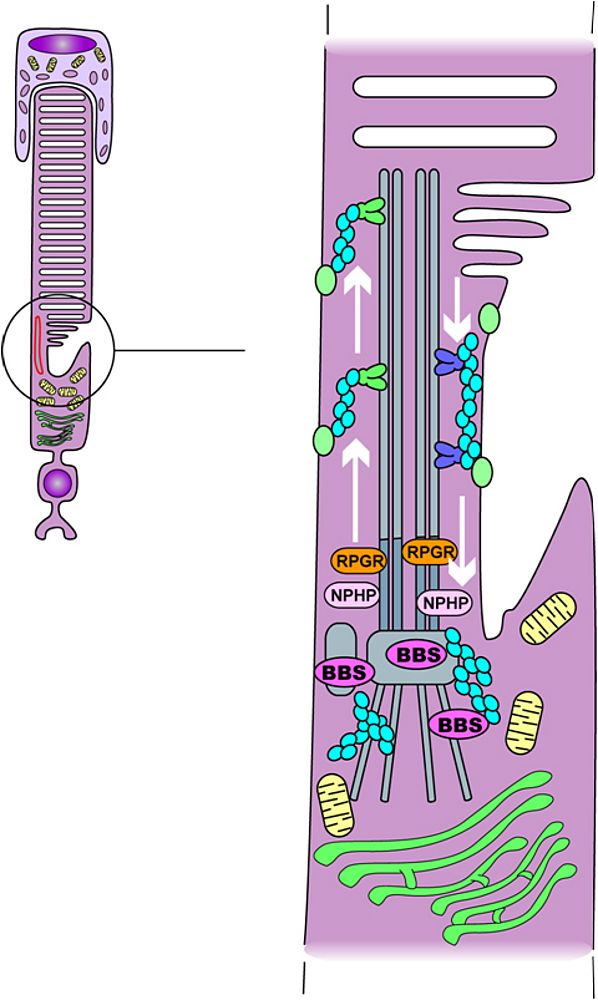 |
*Overview 2: Understanding syndromic retinitis pigmentosa: Bardet-Bield syndrome and the ciliopathies
Primary cilia are found in almost all cells of our body and act as the cell’s antennas capturing extra cellular information (mechanical fluid flow, molecular ligands,…). The photoreceptor cell, with its connecting cilium (located between the outer and the inner segments), is a modified cilium and is of major importance for the trafficking of the phototransduction proteins (translated in the inner segment) to the outer segment. A growing number of proteins are involved in this major cellular process noticeably IFT (Intra Flagellar Transport) proteins and newly recognized proteins involved in the post golgi transport and docking to the cilium system. As a matter of fact, Bardet-Biedl Syndrome (BBS) proteins have been identified by way of gene mapping projects throughout the world including the use of homozygosity mapping. To date, 12 BBS genes have been identified two of which (BBS10 and BBS12) in our laboratory. The challenge is now to understand the action of the latter at the level of the photoreceptor and to prevent retinal degeneration in patients carrying mutations in BBS genes that leads to photoreceptor apoptosis with early onset retinitis pigmentosa as a clinical consequence. To address these challenging questions, we are currently working on in vitro models as well as in vivo models with transgenic mouse models. Meanwhile, 25% of the BBS patients do not show any mutation in any known BBS gene and we are currently searching for new genes to be identified (homozygosity mapping, candidate gene approach, ..).
Inherited eye disorders is to us an emblematic field of research in which the clinical approach is essential to guide the scientific advances. The ultimate goal is to get back to the patients with pathways to therapeutic strategies.
Hélène Dollfus
Inserm - Université de Strasbourg
Hôpitaux Universitaires de Strasbourg