vision-research.eu - The Gateway to European Vision Research
![]()

![]()
You are here: vision-research.eu » Vision Research » Visionary of the Quarter » José-Alain Sahel (Q04-2014)
Gene-independent therapeutic strategies for retinal degeneration
by Prof. José-Alain Sahel
Introduction
 |
Inherited retinal degenerations (IRDs) encompass a large group of clinically and genetically heterogeneous diseases that affect approximately 1 in 3000 people (more than 2 million people worldwide) and represent a major cause of severe and irreversible vision loss. Over the past decade, gene replacement strategies achieved therapeutic success in experimental models of IRDs. Pioneering clinical studies are currently under way to evaluate their safety and efficacy in inherited monogenic eye diseases, such as RPE65-associated Leber congenital amaurosis, Choroideremia, Usher syndrome, Leber hereditary optic neuropathy (LHON) and ABCA4-associated Stargardt disease. We are involved in several of these clinical trials. The extensive genetic heterogeneity and the dominant transmission, however, are major challenges in development of gene therapy for IRDs. Gene-independent strategies could offer promising treatment avenues for these blinding diseases.
Rod-derived Cone Viability Factor (RdCVF)
 |
 |
In many IRDs, mutations selectively affect rod photoreceptors. Secondarily to the death of rods, cones slowly degenerate leading to loss of light-adapted and central vision. This cascade of retinal degenerative events points on the crucial importance of cones in human vision. Saving vision then may come down from prevention of cone degeneration and keeping cone cells alive.
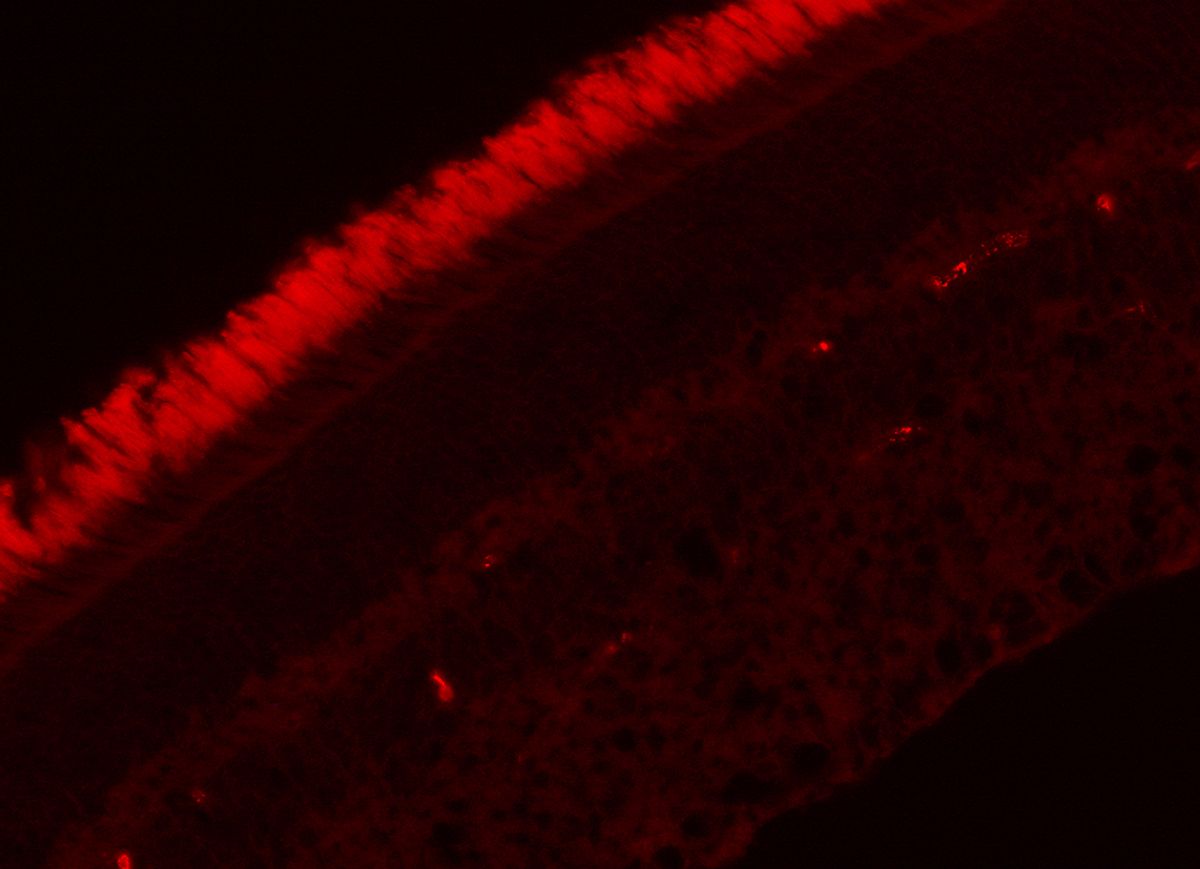
Our group (with Saddek Mohand-Said) hypothesized and demonstrated that rod cells produce a factor that helps the cone cells to survive, and that this factor may preserve vision in human rod-cone dystrophy (retinitis pigmentosa, RP). In the rd1 mouse (an experimental model of RP) we showed that selective transplantation of rods could limit and delay cone loss, and that cone viability depends upon the presence of viable rod photoreceptors. With Thierry Leveillard, screening over two hundred thousand clones (cone-enriched primary cultures from chicken embryos), we identified a protein that increased the number of cone cells in culture and rescued cones in the rd1 mouse retina (Figure 1).
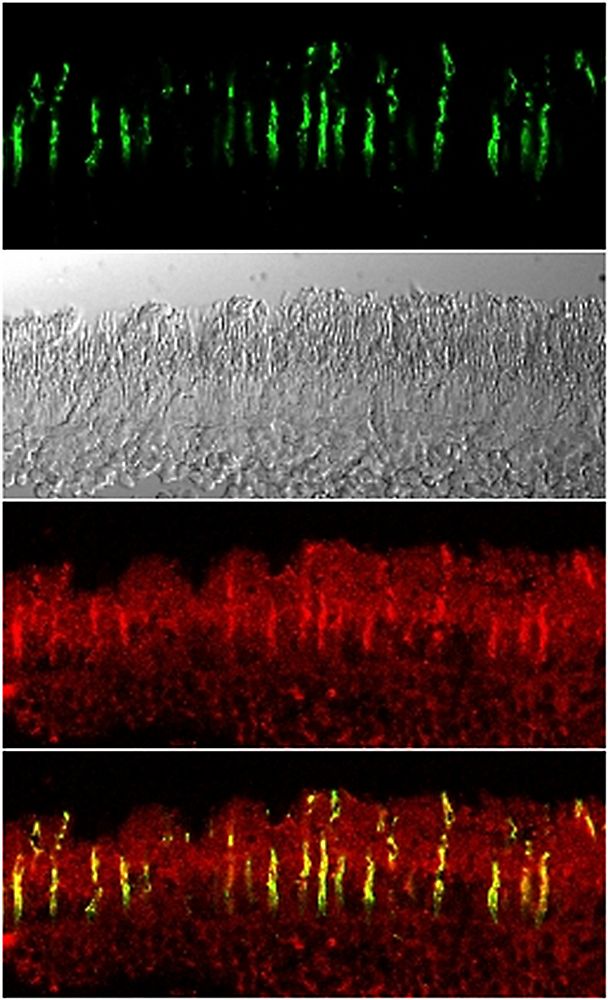
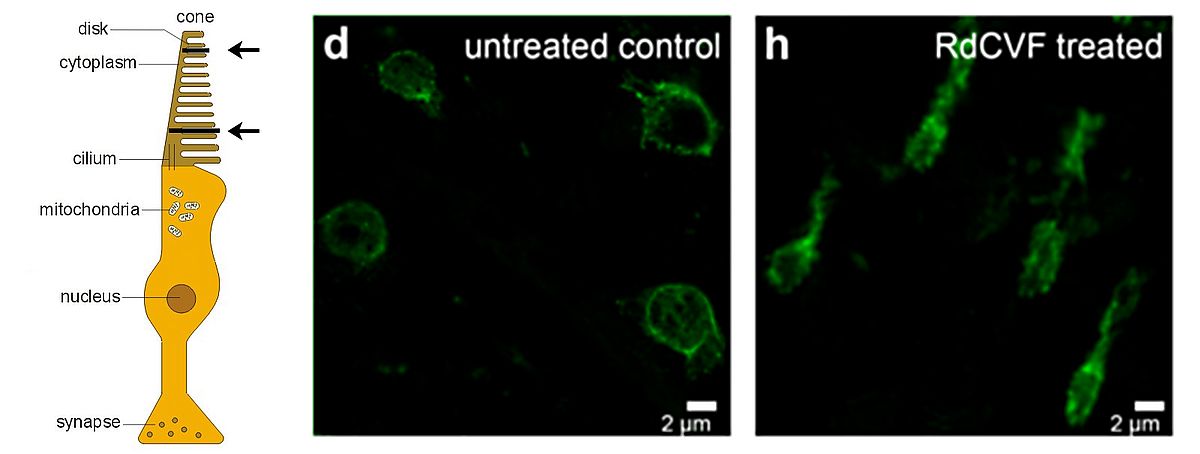
We named this protein Rod-derived Cone Viability Factor (RdCVF) which belongs to the new family of Nucleoredoxin-like (Nxnl1) proteins. We demonstrated that RdCVF administration preserved significantly the cone outer segment (Figure 2) and, correlatively, reduced the loss of function of cone photoreceptors in mutant rodent models. In silico investigations uncovered a second trophic factor belonging to the RdCVF family, RdCVF2 (encoded by the Nxnl2 gene). Similarly to RdCVF, the RdCVF2 short isoform exhibits cone rescue activity. These findings defined a novel gene family expressed in vertebrate retinas with trophic activity and significant potential as therapeutic target in human retinal diseases.
The administration of these survival factors is expected to maintain cones and central vision in most patients suffering from RP. Independent from the disease-causing mutation, RdCVFs shows promise for use in a wide range of IRDs as a neuroprotectant.
 |
Optogenetics for vision restoration
In advanced cases of IRD, there is evidence that cone photoreceptors remain viable with observable cell bodies, for extended periods, but have lost function as a consequence of the loss of outer segments. These "dormant cones" can be converted into “artificial photoreceptors” by targeting their genetically encoded light sensors to elicit retinal activity close to the normal retinal function as proposed and demonstrated by Botond Roska’s group at the Friedrich Miescher Institute for Biomedical Research, Basel, Switzerland, in collaboration with us. The light-activated chloride pump NpHR introduced to surviving cone cell bodies in two mouse models of RP reactivated retinal ON and OFF pathways and retinal circuitry (Figure 3).
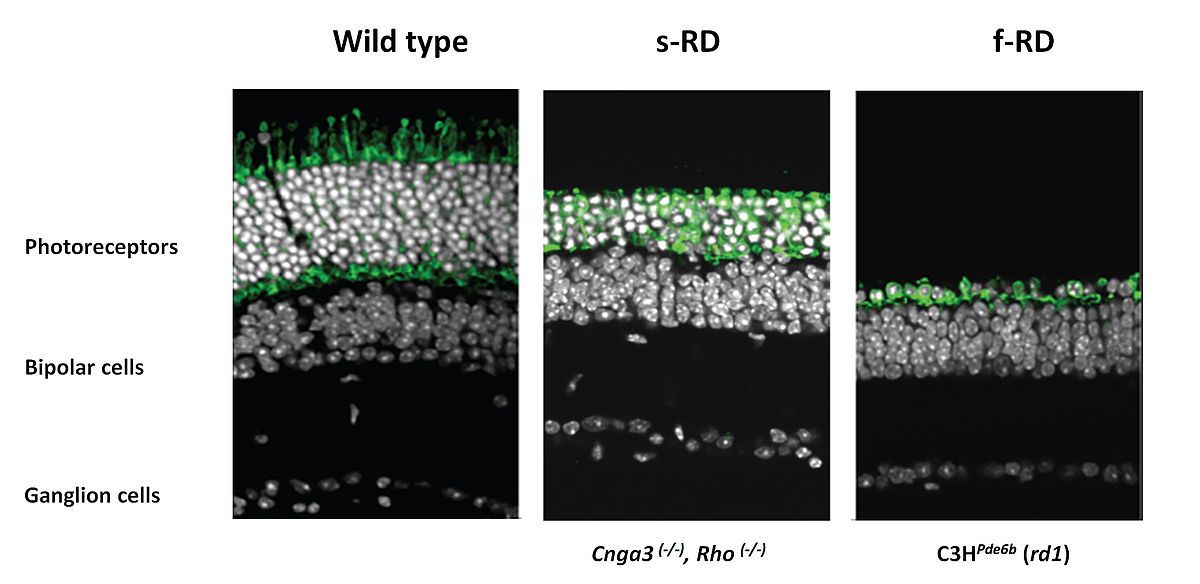 |
Noteworthy, the reactivated cones enabled mice with retinal degeneration to regain visually guided behaviors. Since translation of gene therapy from mice to humans requires the use of promoters and AAV serotypes that drive photoreceptor-specific NpHR expression in human retinas, we tested our AAVs on human ex vivo retinal explants (with Botond Roska, Deniz Dalkara, Jens Duebel and Serge Picaud at Institut de la Vision). For this purpose, we developed an in vitro model system, a post-mortem human retina preparation in culture that allows the investigation of gene expression from viral vectors. One of the vectors used in this study (for which human compatibility has already been demonstrated) was able to express NpHR in cones of human retinal explants. We demonstrated that NpHR targeted to human post-mortem photoreceptors with no measurable intrinsic rod- or cone-mediated photosensitivity restored light responses in photoreceptor cells, clearly demonstrating that reactivation of the surviving retinal structures and phototransduction cascade required for vision is possible. These preclinical data hold promise that legally blind patients or severely visually impaired patients with no visual field but with a preserved layer of cone bodies (visible on optical coherent tomography, OCT) could be eligible for optogenetic functional restoration of cones.
We believe that in addition to IRDs, other blinding diseases such as age-related macular degeneration may also benefit from these developments in the future. Different strategies for optogenetic vision restoration, including their advantages and possible combination with other methods to extend cone cells functional life, to slow retinal degeneration and restore vision were recently reviewed by Sahel and Roska (Ann Rev Neurosci, 2013). This strategy is an attractive alternative to retinal prostheses, a field of active technological, pre-clinical and clinical research for our group and to cell-replacement approaches.
Identifying patients who can benefit from these innovative treatments is a crucial step in the development of therapeutic approaches for retinal degenerative diseases. High-resolution in vivo non-invasive imaging techniques (OCT, adaptive optics) are particularly suited for monitoring the disease progression, establishing functional correlates, studying therapy outcomes and helping to select patients for future clinical trials. The new program HELMHOLTZ, funded by the ERC-Synergy, will, in partnership between the Institut de la Vision and Institut Langevin (Mathias Fink) focus on the development of these high-speed, high resolution 4D tools.
Selected publications
- Macé E, Caplette R, Marre O, Sengupta A, Chaffiol A, Barbe P, Desrosiers M, Bamberg E, Sahel JA, Picaud S, Duebel J, Dalkara D. Targeting Channelrhodopsin-2 to ON-bipolar Cells With Vitreally Administered AAV Restores ON and OFF Visual Responses in Blind Mice. Mol Ther. 2014 Aug 6.
- Reichman S, Terray A, Slembrouck A, Nanteau C, Orieux G, Habeler W, Nandrot EF, Sahel JA, Monville C, Goureau O. From confluent human iPS cells to self-forming neural retina and retinal pigmented epithelium. Proc Natl Acad Sci U S A. 2014 Jun 10;111(23):8518-23.
- Sahel JA, Roska B. Gene therapy for blindness. Annu Rev Neurosci. Ann Rev Neurosci 2013, Jul 8;36:467-88.
- Jaillard C, Mouret A, Niepon ML, Clérin E, Yang Y, Lee-Rivera I, Aït-Ali N, Millet-Puel G, Cronin T, Sedmak T, Raffelsberger W, Kinzel B, Trembleau A, Poch O, Bennett J, Wolfrum U, Lledo PM, Sahel JA, Léveillard T. Nxnl2 splicing results in dual functions in neuronal cell survival and maintenance of cell integrity. Hum Mol Genet. 2012 May 15;21(10):2298-311.
- Sahly I, Dufour E, Schietroma C, Michel V, Bahloul A, Perfettini I, Pepermans E, Estivalet A, Carette D, Aghaie A, Ebermann I, Lelli A, Iribarne M, Hardelin JP, Weil D, Sahel JA, El-Amraoui A, Petit C. Localization of Usher 1 proteins to the photoreceptor calyceal processes, which are absent from mice. J Cell Biol. 2012 Oct 15;199(2):381-99.
- Vignal-Clermont C, Audo I, Sahel JA, Paques M. Poppers-associated retinal toxicity. N Engl J Med. 2010 Oct 14;363(16):1583-5.
- Paques M, Simonutti M, Augustin S, Goupille O, El Mathari B, Sahel JA. In vivo observation of the locomotion of microglial cells in the retina. Glia. 2010 Nov 1;58(14):1663-8.
- Yang Y, Mohand-Said S, Léveillard T, Fontaine V, Simonutti M, Sahel JA. Transplantation of photoreceptor and total neural retina preserves cone function in P23H rhodopsin transgenic rat. PLoS One. 2010 Oct 19;5(10):e13469.
- Busskamp V, Duebel J, Balya D, Fradot M, Viney TJ, Siegert S, Groner AC, Cabuy E, Forster V, Seeliger M, Biel M, Humphries P, Paques M, Mohand-Said S, Trono D, Deisseroth K, Sahel JA, Picaud S, Roska B. Genetic Reactivation of Cone Photoreceptors Restores Visual Responses in Retinitis pigmentosa. Science. 2010 329(5990):413-7.
- Léveillard T, Sahel JA. Rod-derived cone viability factor for treating blinding diseases: from clinic to redox signaling. Sci Transl Med. 2010 Apr 7;2(26):26ps16.
- Yang Y, Mohand-Said S, Danan A, Simonutti M, Fontaine V, Clerin E, Picaud S, Léveillard T, Sahel JA. Functional cone rescue by RdCVF protein in a dominant model of retinitis pigmentosa. Mol Ther. 2009 May;17(5):787-95.
- Léveillard T, Mohand-Saïd S, Lorentz O, Hicks D, Fintz AC, Clérin E, Simonutti M, Forster V, Cavusoglu N, Chalmel F, Dollé P, Poch O, Lambrou G, Sahel JA. Identification and characterization of rod-derived cone viability factor. Nat Genet. 2004 Jul;36(7):755-9.
- Fintz AC, Audo I, Hicks D, Mohand-Said S, Léveillard T, Sahel J. Partial characterization of retina-derived cone neuroprotection in two culture models of photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2003 Feb;44(2):818-25.
- Porto FB, Perrault I, Hicks D, Rozet JM, Hanoteau N, Hanein S, Kaplan J, Sahel JA. Prenatal human ocular degeneration occurs in Leber's congenital amaurosis (LCA2). J Gene Med. 2002 Jul-Aug;4(4):390-6.
- Mohand-Said S, Deudon-Combe A, Hicks D, Simonutti M, Forster V, Fintz AC, Léveillard T, Dreyfus H, Sahel JA. Normal retina releases a diffusible factor stimulating cone survival in the retinal degeneration mouse. Proc Natl Acad Sci U S A. 1998 Jul 7;95(14):8357-62.
Prof. José-Alain Sahel
- Director of the "Institut de la Vision", UPMC-Inserm-CNRS Research Center Paris.
- Chairman, Departments of Ophthalmology: Centre Hospitalier National d'Ophtalmologie des Quinze-Vingts and Rothschild Ophthalmology Foundation, Paris.
- Professor of Ophthalmology Pierre & Marie Curie Medical School, Paris VI Sorbonne Universités.
- Cumberlege Professor of Biomedical Sciences, Institute of Ophthalmology, University College London.
- Académie des Sciences-Institut de France / German National Academy of Sciences Leopoldina / Academia Ophthalmologica Internationalis.
Contact
Prof. José-Alain Sahel
17 rue Moreau
75012 Paris, France
Phone: +33153462513
Fax: +33140021499
E-mail: j.sahel[at]gmail.com