vision-research.eu - The Gateway to European Vision Research
![]()

![]()
You are here: vision-research.eu » Vision Research » Visionary of the Quarter » Carlo Rivolta (Q04-2019)
The major topics of the research work of Professor Carlo Rivolta
 |
Hereditary retinal degenerations (HRDs) are rare diseases that lead to progressive loss of vision and, in many cases, to blindness at the end stage. They invariantly result from genetic mutations that are transmitted from parents to their offspring or that occur spontaneously at the early stages of embryogenesis. In contrast to other rare disorders, for which disease is caused by mutations in a single gene that is also condition-specific, HRDs can be caused by thousands of different mutations in as many as 300 individual genes, and yet result in the same clinical outcome.
As a geneticist, I am interested in identifying such genes and mutations, to better understand the molecular mechanisms leading to retinal blindness. Our typical research project starts from the computer-based analysis of genomic data from families or cohorts of patients with vision impairment and then extends to functional tests at the protein and the cellular levels. My research team is therefore split in two sub-units: one performing in silico analyses on data from Next-Generation Sequencing (NGS), as well as software development, and another working in a wet lab and exploring the molecular genetics of these conditions. In some instances, we also extend our investigations to preclinical studies, by providing support and functional data to other groups involved in gene- or cell-based therapy endeavors.
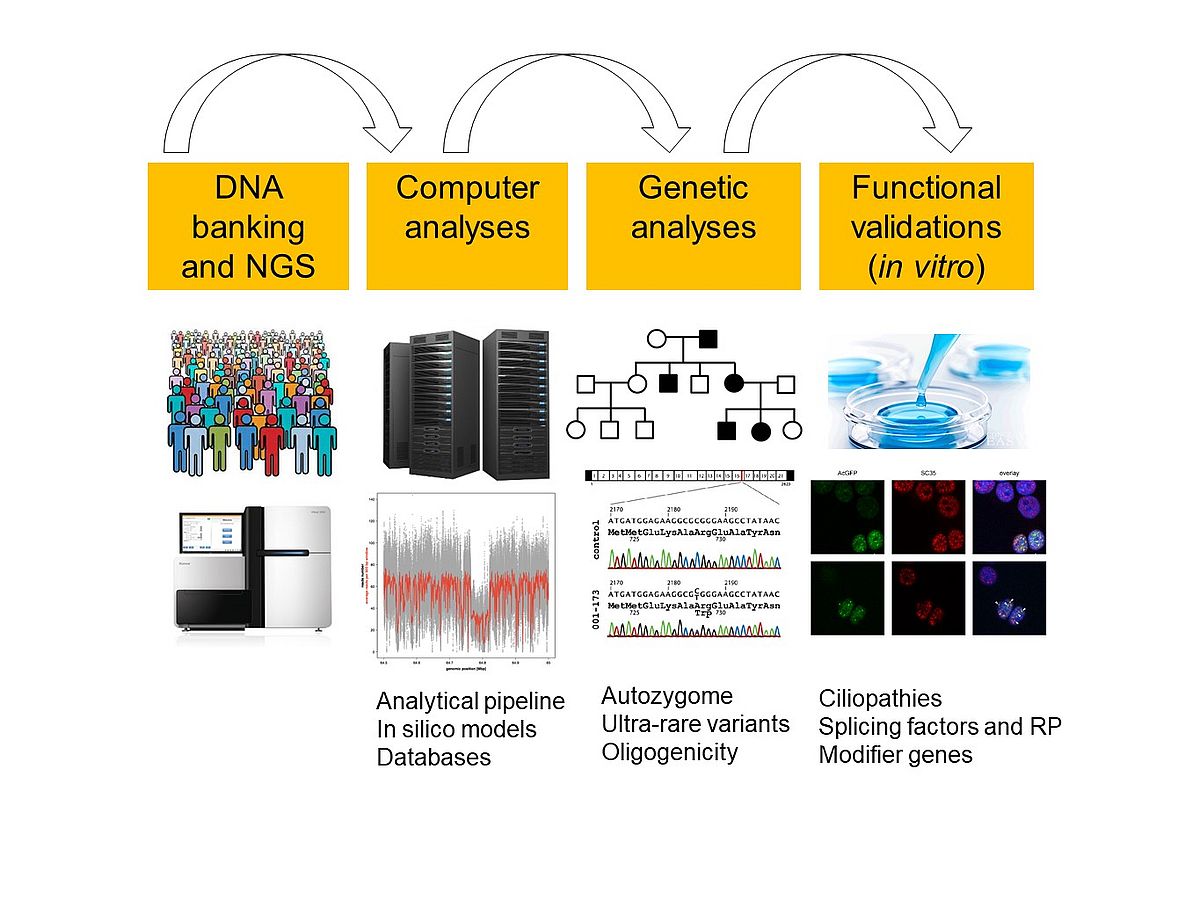 |
Next Generation Sequencing and computer analyses.
Similar to other branches of medical genetics, ophthalmic genetics has been literally revolutionized by the advent of NGS technologies, which have allowed scientists for the first time in history to investigate Gigabases of DNA at once. Unfortunately, the wealth of information produced by NGS represents at the same time a burden for the identification of DNA variants that cause rare diseases, to the point that some patients cannot be diagnosed at the molecular level because of an “excess” of data. Indeed, pathogenic variants typically consist of one or a few mutations that have to be distinguished from thousands of benign DNA changes, genome-wide, and their identification has often been compared to the detection of a needle in a haystack. A part of our research is therefore oriented in developing and constantly refining computer algorithms that could identify and track pathogenic mutations with increasing precision and confidence.
Molecular genetics of HRDs.
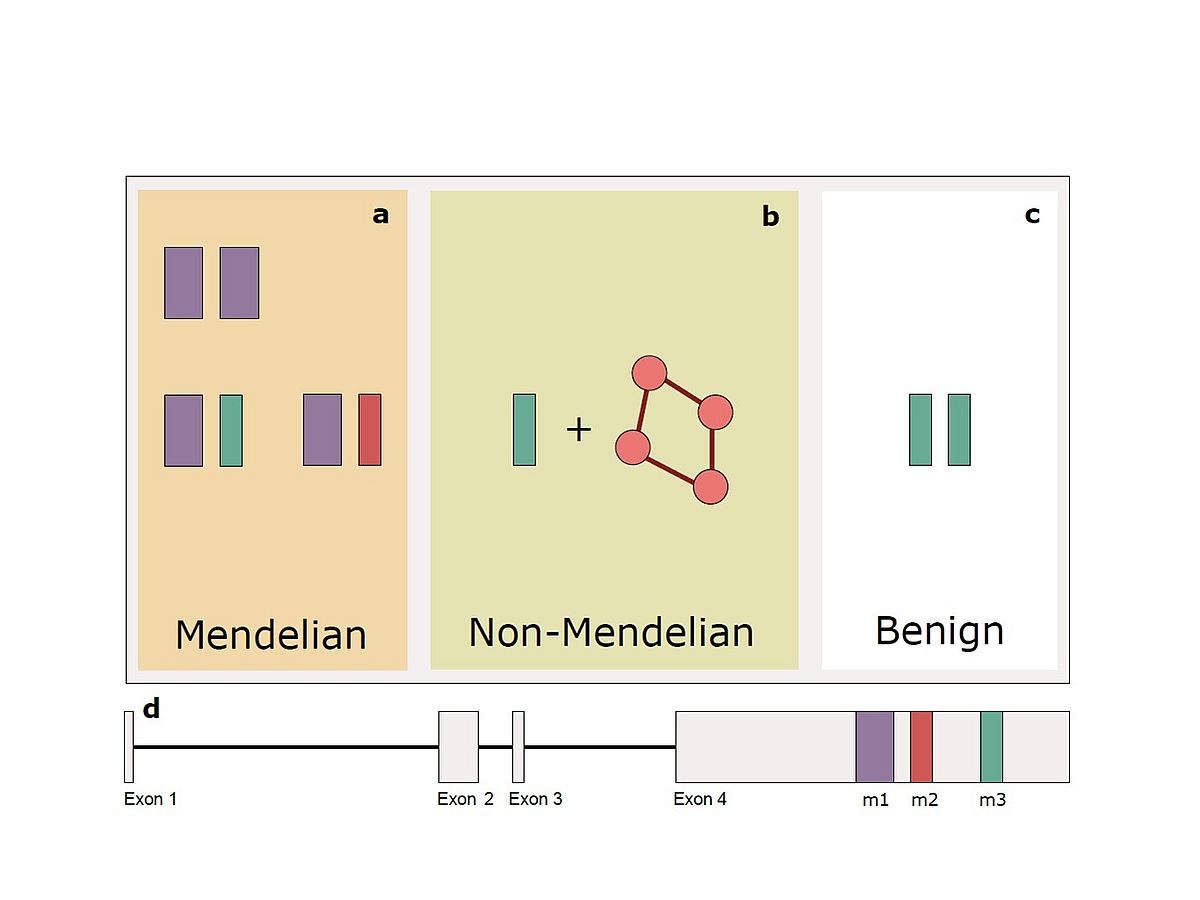
Over the years, the use of NGS on the DNA of relatively large cohorts of patients has allowed us to identify numerous mutations and novel disease genes. However, it has also provided our team a means of analyzing the genetic landscape of these same individuals at a genomic scale. By analyzing such data, we have discovered that for some patients with HRD inheritance of the disease may transcend the monogenic model, and conversely include DNA variants/mutations in multiple genes. We currently ignore the magnitude of this effect at the population level, but we could demonstrate that this is the case for at least one particular DNA variant in the RP1 gene. Since this same variant can behave as a classical, monogenic recessive mutation, an oligogenic allele, or even a benign DNA change, we have termed this particular form of inheritance “quasi-Mendelian”. Along these same lines, we have investigated for a long time the presence and the possible effect on HRD of modifier genes, i.e. of genes that prevent the occurrence of disease in some individuals who nonetheless carry clear-cut mutations. In addition to being useful for the understanding of the molecular mechanisms of pathogenesis, modifier genes may represent a very important element to design therapeutic interventions in patients with specific DNA defects.
 |
Pathomechanisms for novel disease genes.
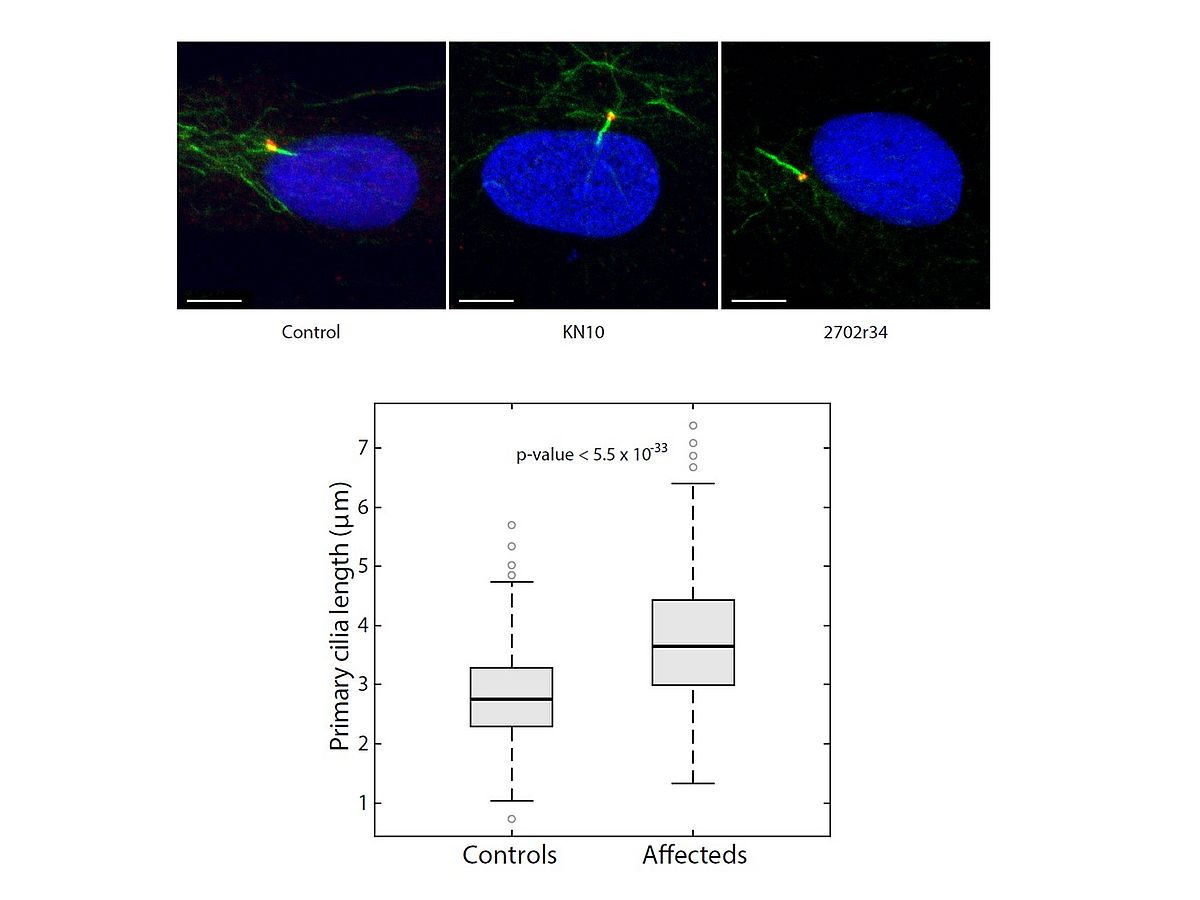
Since our group includes both computer scientists and biologists/physicians, we are in the very privileged position of being able to follow a few projects from NGS analysis to protein characterization. Our specific expertise in terms of in vitro and ex-vivo testing of disease-associated genes involve mainly the characterization of defects leading to HRD through the impairment of the splicing process (mutations in the PRPF genes) or affecting the homeostasis of photoreceptors’ connecting cilium, determining the broad class of retinal ciliopathies.
 |
References
- Nikopoulos K, Cisarova K, Quinodoz M, Koskiniemi-Kuendig H, Miyake N, Farinelli P, Rehman AU, Khan MI, Prunotto A, Akiyama M, Kamatani Y, Terao C, Miya F, Ikeda Y, Ueno S, Fuse N, Murakami A, Wada Y, Terasaki H, Sonoda KH, Ishibashi T, Kubo M, Cremers FPM, Kutalik Z, Matsumoto N, Nishiguchi KM, Nakazawa T, Rivolta C. (2019). A frequent variant in the Japanese population determines quasi-Mendelian inheritance of rare retinal ciliopathy. Nat Commun 10: 2884.
- Quinodoz M, Royer-Bertrand B, Cisarova K, Di Gioia SA, Superti-Furga A, Rivolta C. (2017). DOMINO: using machine-learning to predict genes associated with dominant disorders. Am J Hum Genet. 101:623-629.
- Bedoni N, Haer-Wigman L, Vaclavik V, Tran V, Farinelli P, Balzano S, Royer-Bertrand B, El-Asrag M, Bonny O, Ikonomidis C, Litzistorf Y, Nikopoulos K, Yioti GG, Stefaniotou MI, McKibbin M, Booth AP, Ellingford JM, Black GC, Toomes C, Inglehearn CF, Hoyng CB, Bax N, Klaver CCW, Thiadens AA, Murisier F, Schorderet DF, Ali M, Cremers FPM, Andreasson, S Munier FL, Rivolta C. (2016). Mutations in the polyglutamylase gene TTLL5, expressed in photoreceptor cells and spermatozoa, are associated with cone-rod degeneration and reduced male fertility. Hum Mol Genet. 25:4546-4555.
- Nikopoulos K, Farinelli P, Giangreco B, Tsika C, Royer-Bertrand B, Mbefo MK, Kjellstrom U, El Zaoui I, Di Gioia SA, Balzano S, Cisarova K, Messina A, Decembrini S, Plainis S, Mplazaki S, Khan MI, Michael S, Boldt K, Ueffing M, Moulin AP, Cremers FPM, Roepman R, Arsenijevic Y, Tsilimbaris MK, Andreasson S, Rivolta C. (2016). Mutations in CEP78 cause cone-rod dystrophy and hearing loss associated with primary cilia defects. Am J Hum Genet. 99:770-6.
- Di Gioia SA, Farinelli P, Letteboer SJ, Arsenijevic Y, Sharon D, Roepman R and Rivolta C. (2015). Interactome analysis reveals that FAM161A, deficient in recessive retinitis pigmentosa, is a component of the Golgi-centrosomal network. Hum Mol Genet 15: 3359-3371.
- Nishiguchi KM, Tearle RG, Liu YP, Oh EC, Miyake N, Benaglio P, Harper S, Koskiniemi-Kuendig H, Venturini G, Sharon D, Koenekoop RK, Nakamura M, Kondo M, Ueno S, Yasuma TR, Beckmann JS, Ikegawa S, Matsumoto N, Terasaki H, Berson EL, Katsanis N and Rivolta C. (2013). Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc Natl Acad Sci U S A. 110:16139-16144.
- Venturini G, Rose AM, Shah AZ, Bhattacharya SS and Rivolta C. (2012). CNOT3 is a modifier of PRPF31 mutations in retinitis pigmentosa with incomplete penetrance. PLoS Genet 8: e1003040.
- Di Gioia SA, Letteboer SJ, Kostic C, Bandah-Rozenfeld D, Hetterschijt L, Sharon D, Arsenijevic Y, Roepman R and Rivolta C. (2012). FAM161A, associated with retinitis pigmentosa, is a component of the cilia-basal body complex and interacts with proteins involved in ciliopathies. Hum Mol Genet 21:5174-5184.
- Tanackovic G, Ransijn A, Ayuso C, Harper S, Berson EL and Rivolta C (2011). A missense mutation in PRPF6 causes impairment of pre-mRNA splicing and autosomal dominant retinitis pigmentosa. Am J Hum Genet. 88:643-649.
- Tanackovic G, Ransijn A, Thibault P, Abou Elela S, Klinck R, Berson EL, Chabot B and Rivolta C (2011). PRPF mutations are associated with generalized defects in spliceosome formation and pre-mRNA splicing in patients with retinitis pigmentosa. Hum Mol Genet 20:2116-21130.

Professor Carlo Rivolta
Institute of Molecular and Clinical Ophthalmology Basel (IOB)
Mittlere Strasse 91,
CH-4031 Basel,
Switzerland
E-mail: carlo.rivolta[at]iob.ch
Website: https://www.iob.ch/