vision-research.eu - The Gateway to European Vision Research
![]()

![]()
You are here: vision-research.eu » Vision Research » Visionary of the Quarter » Valeria Marigo (Q04-2021)
The research work of Professor Valeria Marigo
 |
Inherited retinal degeneration (IRD) is a group of diseases characterized by progressive loss of photoreceptor cells, the cells in the eye that respond to light, and this damage affects vision and ultimately leads to complete blindness1. IRD comprises retinitis pigmentosa (RP) and Leber congenital amaurosis (LCA) and can be caused by mutations in more than 100 different genes. This high genetic heterogeneity has hampered the development of therapeutic interventions.
The linkage of genes to IRD, the identification of mutations in IRD patients and the characterization of molecular events underlying the retinal degenerative process provided ample knowledge on the causes of the disease. Based on data collected on cellular and animal models we think that the molecular pathways disturbed during photoreceptor degeneration represent possible targets for neuroprotective therapeutic strategies.
The approved successful therapy for LCA type 2, LCA2, is based on gene therapy, which targets the retinal pigment epithelium replacing the mutated RPE65 gene 2. Nonetheless, such type of therapies, engineered for specific genes that cause an individual form of IRD, can reach very few patients and it is unlikely that such a personalized therapy will ever be available for all IRD patients. Moreover, most gene therapy trials are gene replacing treatments, appropriate only for recessive forms of retinal degeneration 3. Hence, there is a therapeutic need for the development of treatments that can reach a high number of patients bearing mutations in different genes, overcoming the setback derived from the genetic heterogeneity of IRD and the type of hereditary trait. Neuroprotection is an option that we are evaluating in the lab to target common molecular denominators of the degeneration process, because it holds high potentials for treatment of a broader cohort of patients.
In the last decade, we and others characterized the pathophysiological events occurring at the subcellular and molecular levels in degenerating photoreceptors. While there is a great functional diversity in the genes linked to IRD, some common denominators have been identified that play key roles during the degenerative process. An early alteration in several murine models of RP and LCA is the increase in intracellular levels of cGMP 4. High cGMP in photoreceptors causes influx of calcium ions and activation of calpain proteases and protein kinase G (PKG) (Figure 1).
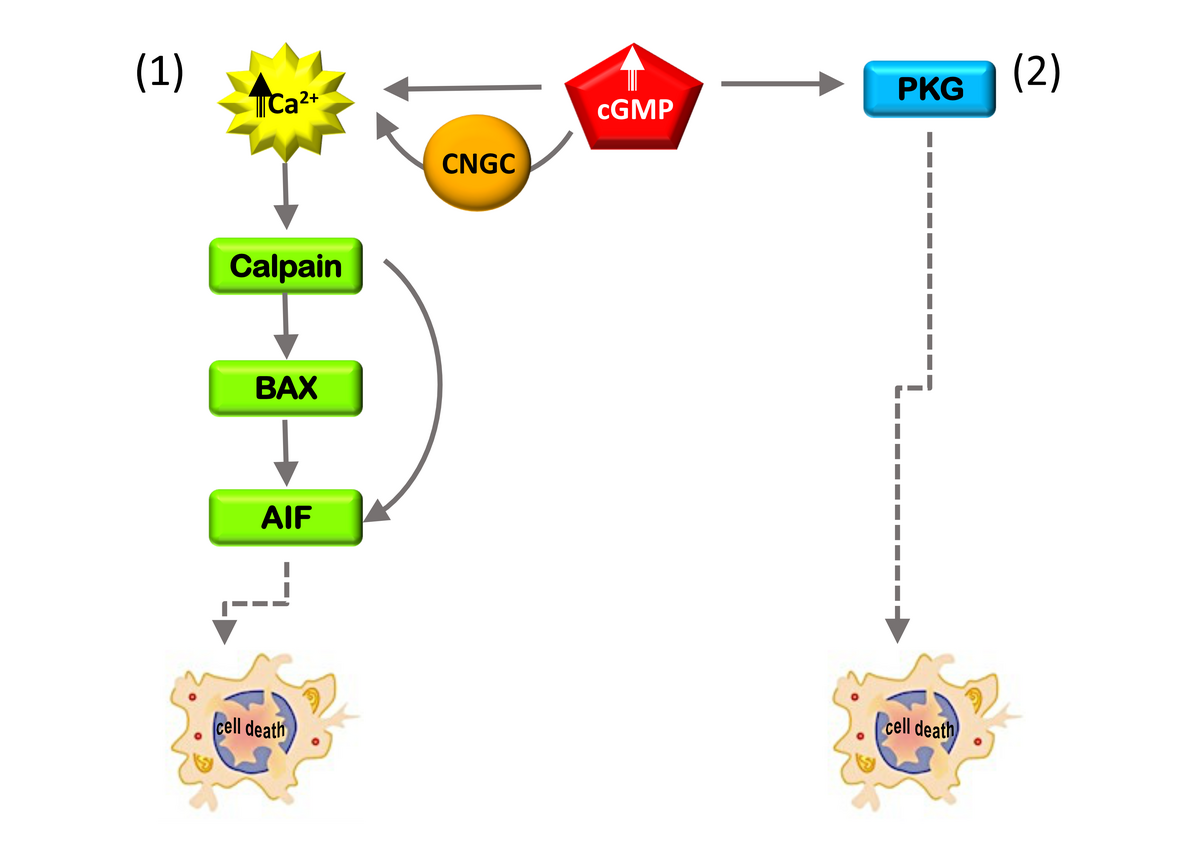 |
The questions that we are addressing at the moment are: “how can we target these pathways to restrain retinal degeneration?” and “considering that the progression of the disease can be very slow, how can we deliver the neuroprotective molecule for a long-lasting effect?”.
In a collaborative effort funded by several EU projects, we identified a cGMP analogue with an inhibiting activity, thus able to counteract the excess of cGMP inside dying photoreceptors. This compound demonstrated neuroprotective activity, by decreasing the number of dying photoreceptors, in a cell model of the disease, in organ explants from murine models and in vivo in mice bearing mutations causing RP 5. Next step will be developing an appropriate delivery system for favourable pharmacokinetics and pharmacodynamics in a long-lasting supply of the inhibiting compound to the degenerating retina 6.
A second approach targets excess intracellular calcium, a common feature of dying retinal neurons. The pigment epithelium-derived factor (PEDF) is a natural protein expressed in the healthy human eye and its levels are altered in eyes affected by RD 7. In collaboration with Dr Patricia Becerra at NEI we delivered intravitreally the full protein and of a small peptide, only 17 amino acids, of PEDF in RP murine models and confirmed that the recombinant protein has neuroprotective properties and can delay photoreceptor cell death 8. We also defined that PEDF acts on calcium pumps to decrease intracellular calcium levels (Figure 2) 9.
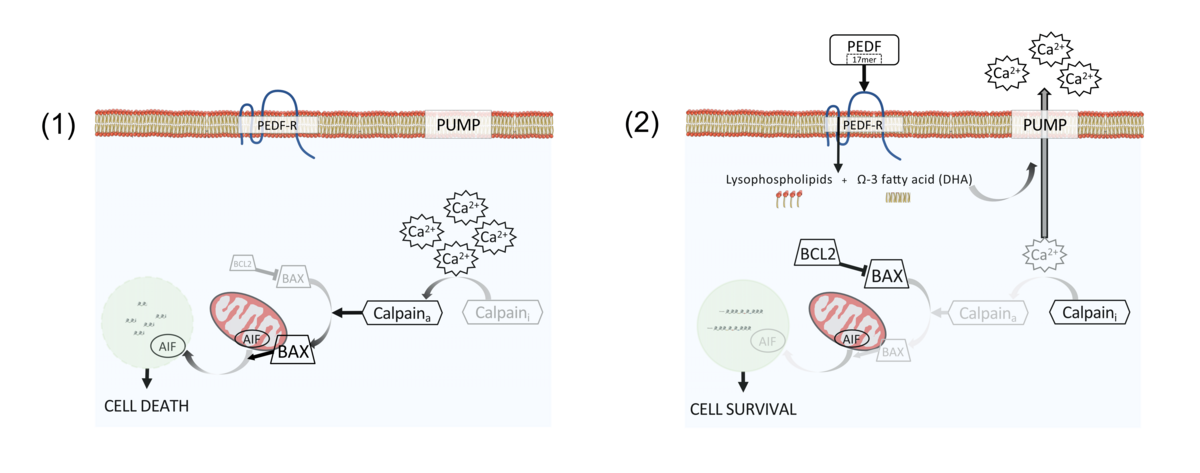 |
We are now developing a new biotechnological delivery system for sustained exposure of the degenerating retina to PEDF or to the active 17 amino acids peptide.
The progression of neurodegenerative diseases can be exceedingly slow, with often decades until the manifestation of overt symptoms, in spite of the disease relentlessly advancing. This feature leaves a wide window of intervention for neuroprotective treatments and the collaboration with clinicians in ophthalmology will be fundamental to translate our research from bench to patients.
Bibliography
- Broadgate, S., Yu, J., Downes, S. M. & Halford, S. Unravelling the genetics of inherited retinal dystrophies: Past, present and future. Progr Retin Eye Res 59, 53–96 (2017).
- Trapani, I. & Auricchio, A. Seeing the Light after 25 Years of Retinal Gene Therapy. Trends Mol Med 24, 669–681 (2018).
- Trapani, I. & Auricchio, A. Has retinal gene therapy come of age? From bench to bedside and back to bench. Hum Mol Genet 28, R108–R118 (2019).
- Power, M. et al. Cellular mechanisms of hereditary photoreceptor degeneration – Focus on cGMP. Progr Retin Eye Res 74, 100772 (2020).
- Vighi, E. et al. Combination of cGMP analogue and drug delivery system provides functional protection in hereditary retinal degeneration. Proc Natl Acad Sci USA 115, E2997–E3006 (2018).
- Himawan, E. et al. Drug delivery to retinal photoreceptors. Drug Discov Today 24, 1637–1643 (2019).
- Ogata, N., Matsuoka, M., Imaizumi, M., Arichi, M. & Matsumura, M. Decreased levels of pigment epithelium-derived factor in eyes with neuroretinal dystrophic diseases. Am J Ophthalmol 137, 1129–1130 (2004).
- Kenealey, J. et al. Small Retinoprotective Peptides Reveal a Receptor-binding Region on Pigment Epithelium-derived Factor. J Biol Chem 290, 25241–25253 (2015).
- Comitato, A. et al. Pigment epithelium-derived factor hinders photoreceptor cell death by reducing intracellular calcium in the degenerating retina. Cell Death Dis. 9, 560 (2018).

Professor Valeria Marigo
Full professor of Molecular Biology,
Head of the bachelor school of Biotechnology
Department of Life Sciences,
University of Modena and Reggio Emilia,
via G. Campi, 287
41125 Modena,
Italy
E-mail: vmarigo[at]unimore.it