vision-research.eu - The Gateway to European Vision Research
![]()

![]()
You are here: vision-research.eu » Vision Research » Visionary of the Quarter » Robin Ali (Q01-2012)
Gene and stem cell therapy for retinal disorders
 |
The eye has several features that make it particularly suitable as a target organ for the development of novel therapies. The transparent nature of ocular tissues enables the visualization of the delivery reagents and subsequent non-invasive imaging. It is a small, compartmentalized and enclosed organ, enabling the delivery of small quantities of vector to particular ocular compartments with minimal risk of systemic vector dissemination. The blood-retinal barrier and blood-aqueous barrier maintain a degree of protection from immune responses that might otherwise cause inflammation and limit transgene expression. Finally, retinal function can be assessed using a variety of electrophysiological and psychophysical tests commonly used in the clinic.
Gene therapy
Gene therapy is a powerful new approach for the treatment of eye disease. We aim to develop gene therapy not only for a number of relatively rare, but currently untreatable, inherited retinopathies, but also to develop more effective treatments for common ocular disorders involving neovascularisation and inflammation. Gene therapy offers the prospect of local treatment with reduced risk of systemic side-effects and the potential for long term efficacy following a single administration of vector that could be much more cost-effective than repeated drug administration.
Major advances in understanding disease processes and the development of efficient gene transfer to the eye using adeno-associated virus (AAV) and lentiviral vectors have led to robust proof-of-concept studies of gene therapy for both inherited and complex disorders of the eye using a variety of animal models. We have been working for the past two decades in this rapidly growing field. Our achievements include the first demonstration of efficient photoreceptor transduction (1), the first demonstration of functional rescue of mouse model of retinitis pigmentosa that established proof-of-concept for ocular gene therapy (2), the development of various viral vector systems for use in eye, including a non-integrating lentiviral vector (3) and in 2007, the establishment of the world’s first clinical trial of gene therapy for inherited retinopathy. The first set of results from this trial (4) reporting an improvement in vision in patients with Leber congential amaurosis type 2 (LCA2), along with results from two other clinical trials have established proof-of-principle of gene therapy for inherited retinal degeneration (http://news.bbc.co.uk/1/hi/health/7370694.stm).
We have established a translational pipeline to take gene therapy from proof-of-concept in animal models through to clinical trials and clinical application. Together with our partners at University College London, we have developed a pilot facility for manufacture of AAV and lentiviral vectors. This facility has been recently accredited by MHRA and is now being used to manufacture clinical grade gene therapy vectors for use in our clinical trials. We have also established a core team of experienced research staff who can co-ordinate the pre-clinical safety and efficacy studies, develop trial protocols and prepare submissions to the regulatory authorities.
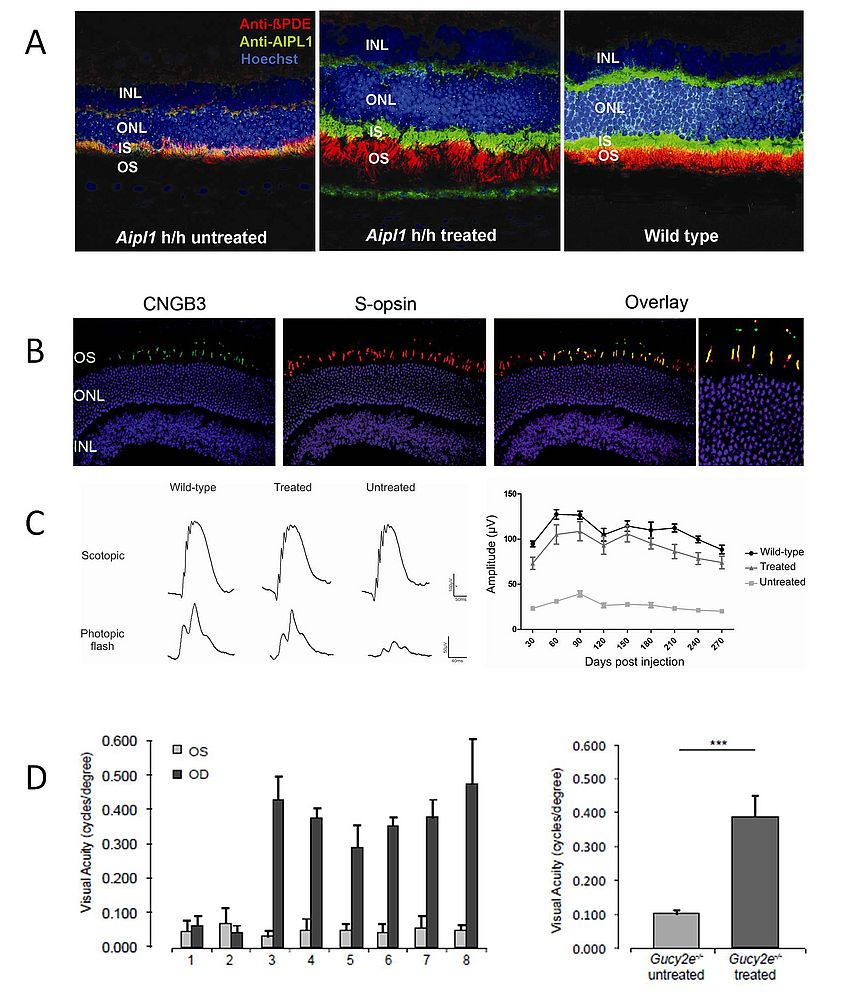
Figure 1: Gene supplementation therapy leads to improved function in rod and cone photoreceptor cells
- Subretinal administration of AAV2.AIPL1 in Aipl1 hypomorphic mice restores AIPL1 protein (green) in the rod photoreceptors and rescues the translocation of phosphodiesterase (PDE, red), a client protein of AIPL1.
- The cone arrestin promoter (CAR) drives cone specific expression of the CNGB3 gene (green) in Cngb3 knock-out mice after subretinal injection of AAV2/8.CAR.CNGB3, as shown by co-localisation with the cone S-opsin (red).
- CNGB3 gene supplementation therapy rescues the photopic electroretinogram (ERG) to near wild type level 4 months post-treatment (left). ERG rescue after treatment is preserved for at least 9 months (right).
- Transfer of the GUCY2D gene to the rods and cones of knock-out mice using an AAV2/8 vector leads to an improvement in visual acuity, assessed by photopic optomotor responses. Animals 1 and 2 were untreated knock-out mice. Of animals 3-8 only the left eye (OD) was treated. The graph on the right shows average acuities of treated and untreated eyes 4 months post-injection.
Gene therapy for inherited retinal degenerations
We are now building on the experience we have gained in our first clinical trial to develop gene therapy for other severe inherited retinal degenerations. Since the majority of inherited retinal degenerations result from mutations in photoreceptor-specific genes, proof of principle of gene therapy for a photoreceptor-specific form of LCA would expand significantly the range of retinal disorders potentially amenable to this approach. In recent years, we have been able to demonstrate proof-of-concept for gene therapy treatment in animal models of a number of other forms of LCA that are caused by mutations in photoreceptor genes. These include LCA4 - AIPL1 deficiency - (5), LCA1 - GUCY2D deficiency - (6), LCA6 - RPGRIP deficiency - (7) and RP38 - Mertk deficiency- (8). We are currently in the process of developing gene therapy approaches for other forms of LCA including LCA10 (CEP290 deficiency), LCA13 (RDH12 deficiency; in collaboration with Prof D Thompson, Michigan, USA) and LCA8 (CRB1 deficiency; in collaboration with Prof J Wijnholds, Amsterdam, The Netherlands). In addition, we have performed robust proof of concept studies of gene replacement therapy for the most common form of retinitis pigmentosa, X-linked RP3, RPGR deficiency, (manuscript in preparation), and for the most prevalent form of achromatopsia, achromatopsia-3, CNGB3 deficiency (9). In this way we have established a therapeutic pipeline for at least 11 forms of early onset severe retinal dystrophies including 9 of the16 known forms of LCA. On the basis of their specific disease features and their responses to gene therapy in animal models we have identified four conditions that we believe to be particularly suitable for clinical trials based on severity of disease, numbers of patients and efficacy of therapy in animal models. Of the four novel therapies, the project for LCA4 is advanced furthest towards human application in a clinical trial.
Gene therapy for the treatment of ocular angiogenesis
Angiogenic diseases of the retina include age related macular degeneration (AMD) and diabetic retinopathy and are among the leading causes of visual impairment in the developed world. Their global prevalence is increasing as a result of demographic changes and the diabetes epidemic. The currently available treatments Lucentis and Avastin (off-licence) target vascular endothelial growth factor (VEGF) non-specifically. Currently available agents can help control neovascularisation in forms of AMD and diabetes, but are dependent on invasive monthly intravitreal injections making them very expensive to administer. Sustained local delivery by vector-mediated expression therefore offers a major advantage over repeated intraocular injection. We published the first proof-of-concept studies that demonstrated the utility of AAV-mediated delivery of sFlt1 (10) and lentiviral-mediated delivery of endostatin and angiostatin (11) for the treatment of ocular neovascularisation. We are currently pursuing the development of more effective gene therapy vectors that will enable sustained local delivery of various angiostatic molecules with enhanced specificity.
Gene therapy for the treatment of ocular inflammation
We are also developing gene therapy approaches to immune-mediated ocular diseases such uveitis and have demonstrated that intraocular inflammation can be suppressed by AAV- or lentiviral vector-mediated expression of the immunosuppressive cytokine IL-10, a cytokine expressed in the retina to maintain homeostasis (12, 13). Underpinning this translational drive is a pipeline of other targets including FasL and TGFb for which we have also demonstrated that vector-mediated delivery suppresses inflammation and restores tissue homeostasis in both uveitis and corneal graft rejection. The added value of this approach is the link with inflammation that is also central to the pathogenesis of AMD. The ability to modulate inflammation in the eye will also inform our underpinning laboratory research into using gene therapy approaches to modulate inflammation as a strategy for the treatment of AMD.
Stem cell therapy
Most forms of untreatable blindness result from the death of retinal neurons. The loss of retinal ganglion cells causes blindness in glaucoma, whereas photoreceptor cell death is the main cause of vision loss in AMD, diabetic retinopathy and inherited retinal degenerations. The first gene therapy trials for retinal disease have set the scene for pioneering new therapies for retinal disease. However, gene therapy approaches are most suitable for treating disorders before the degenerative process has resulted in extensive retinal cell loss and there are many retinal disorders that are not currently amenable to gene therapy. An alternative approach is to replace lost photoreceptors or ganglion cells using cell transplantation. Although ganglion cell replacement might be possible in the future, appropriate regeneration of the long axonal projections to the brain presents a major challenge that has not yet been addressed. Transplantation of photoreceptor cells, however, has shown considerably more promise and is the focus of our cell therapy programme.
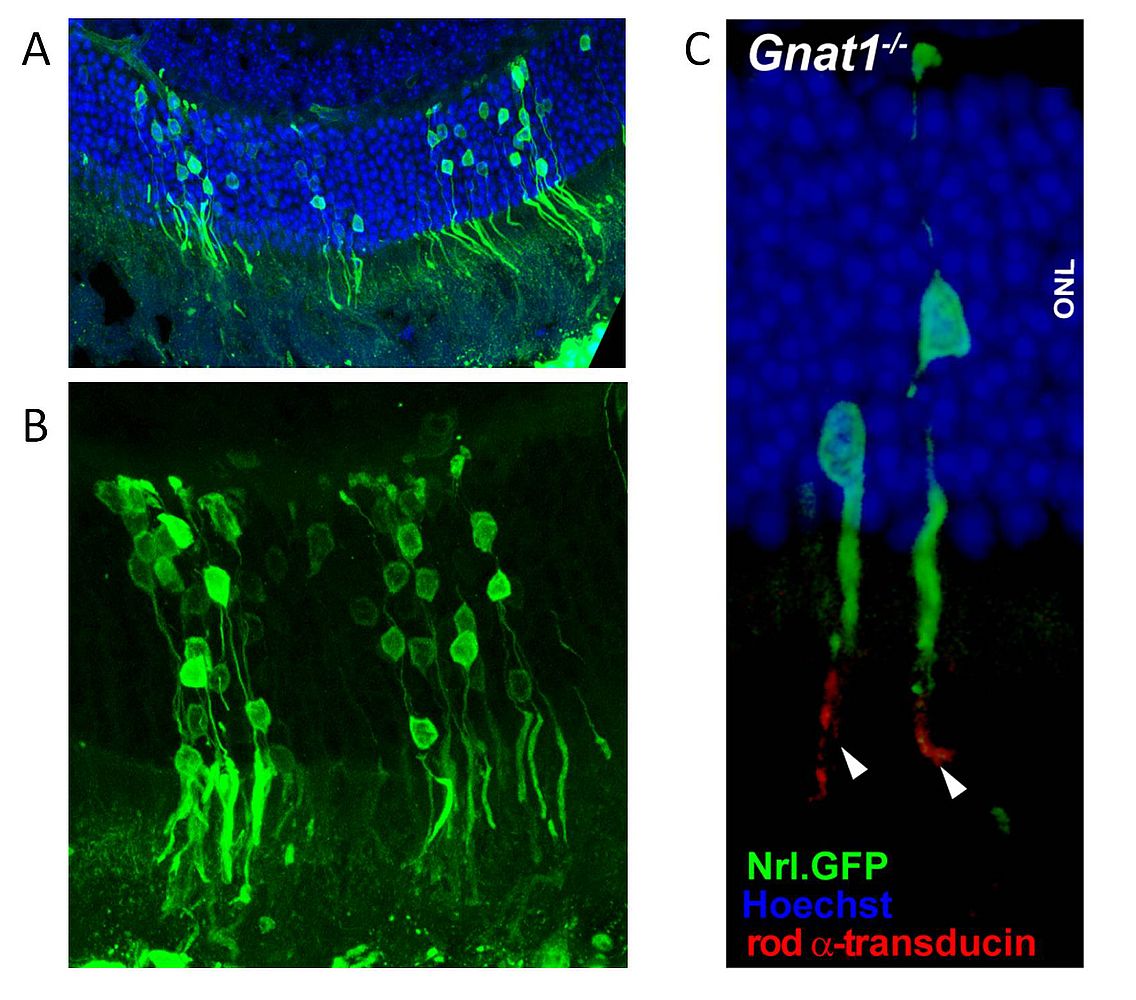
Figure 2: Integration of transplanted photoreceptor cell precursors into adult recipient retinae
- Overview of an area of wild type murine retina with GFP labelled donor photoreceptor cells 4 weeks after subretinal administration of photoreceptor precursors isolated from P5 donor retina.
- Integrated donor photoreceptor cells show GFP positive cell bodies, inner segments and synapses, connected by photoreceptor processes.
- High magnification image of integrated photoreceptor cells in a Gnat1 -/- recipient retina. Immunohistochemistry shows that only transplanted photoreceptor cells are positive for Gnat1 protein (rod transducin, white arrowheads).
Rod photoreceptor transplantation
In 2006 we made a significant breakthrough by identifying a defined donor cell population of post-mitotic rod precursors from the developing neural retina, which can form new functional rod photoreceptors after transplantation in the adult diseased mouse retina (14). This discovery provided strong justification for the development of stem cell therapy for retinal disease. Over the last five years we have built a comprehensive programme of work to develop stem cell therapy for retinal repair. To date we have focused on developing rod photoreceptor transplantation: We have shown that we can increase the efficacy of transplantation by modulating the retinal environment either by immune suppression (15), modulation of physical barriers (16; 17) or up-regulation of growth factors (West et al., Cell Transplantation, in press). We are also investigating the process by which post-mitotic rod precursors migrate and integrate (Warre-Cornish et al., in preparation) and have examined the impact of disease type and stage on transplantation efficiency in a variety of models of retinal degeneration; the success of transplantation varies between models and is dependent on the rate of degeneration and degree of gliosis (Barber et al., submitted for publication).
We have also shown that so-called "retinal stem cells" from the ciliary margin of the mammalian retina and other adult stem cell sources have limited potential for generating photoreceptors compared with embryonic stem (ES) cells (18; 19). We have therefore focused our efforts on differentiating mouse and human ES cells. In collaboration with Dr M Takahashi and Dr Y Sasai (Riken Institute, Kobe, Japan), we have established highly effective techniques for ES cell differentiation, based on their protocols. In collaboration with the Riken group, we have generated ES cells expressing genetic markers that allow us to direct differentiation by first selecting retinal progenitors and then differentiating these into rod precursors. We will now determine whether ES cell-derived rod precursors are as effective for transplantation as those isolated from the developing neural retina. We have also identified cell surface markers that enable us to select precursor cells for transplantation that are not genetically labelled (20). This is essential for translation to humans.
Our main achievement over the last four years has been to improve the efficiency of rod precursor transplantation to such an extent that we have been able to provide definitive evidence of restoration of rod-mediated visually guided behavior in rod-deficient mice following transplantation. This establishes a major proof of concept - that photoreceptor transplantation has the potential to improve not only retinal function but actually restore vision. (Pearson et al, submitted for publication).
Cone photoreceptor transplantation
To date we have focused on rod transplantation in models of rod degeneration in order to establish the principles of effective photoreceptor transplantation. As humans rely primarily on cone and not rod vision, cone transplantation is likely to be the most effective strategy for clinical application. Cones function in bright (photopic) light, while rods function in very dim (scotopic) light becoming bleached in bright light. In late stage retinitis pigmentosa (RP), despite loss of the majority of their rods, patients retain useful vision until the last remaining cones degenerate. Since the mammalian retina consists of only three to five per cent cones (three per cent in mice, five per cent in humans) we can expect to restore significant function with the replacement of relatively few cones.
We have recently demonstrated proof-of-principle for cone transplantation (21) and have shown that the optimal ontogenetic stage for cone transplantation is similar to that for rods i.e. post-mitotic precursors. Although the adult retinal environment favors rod integration, low numbers of cones can integrate. We now aim to manipulate the developmental mechanisms that limit cone integration and use our acquired knowledge of how to effectively isolate and transplant precursors in order to achieve optimal integration of functional cones. We are seeking to prove the concept that it is possible to restore photopic vision to mice by cone transplantation based on our proven strategy for rods.
References
- Gene transfer into the mouse retina mediated by an adeno-associated viral vector.
RR Ali, MB Reichel, AJ Thrasher, RJ Levinsky, C Kinnon, N Kanuga, DM Hunt and SS Bhattacharya
Human Mol. Genet. 1996; 5 (5): 591-594 - Restoration of photoreceptor ultrastructure and function in retinal degeneration slow mice by gene therapy.
RR Ali, G Sarra, C Stephens, M de Alwis, JWB Bainbridge, PM Munro, S Fauser, MB Reichel, C Kinnon, DM Hunt, SS Bhattacharya and AJ Thrasher
Nat. Genet. 2000; 25 (3): 306-310 - Effective gene therapy with non-integrating lentiviral vectors.
RJ Yáñez-Muñoz, KS Balaggan, A MacNeil, S Howe, M Schmidt, AJ Smith, P Buch, RE MacLaren, PN Anderson, SE Barker, Y Durán, C Bartholomae, C Von Kalle, JR Heckenlively, C Kinnon, RR Ali and AJ Thrasher
Nat. Med. 2006; 12 (3) :348-353 - Effect of gene therapy on visual function in Leber Congenital Amaurosis.
JWB Bainbridge, AJ Smith, SE Barker, S Robbie, R Henderson, KS Balaggan, A Viswanathan, GE Holder, A Stockman, N Tyler, S Petersen-Jones, SS Bhattacharya, AJ Thrasher, FW Fitzke, BJ Carter, GS Rubin, AT Moore and RR Ali
N Engl J Med. 2008; 358 (21): 2231-9 - Gene therapy for retinitis pigmentosa and Leber congenital amaurosis caused by defects in AIPL1: effective rescue of mouse models of partial and complete Aipl1 deficiency using AAV2/8 vectors.
MH Tan, AJ Smith, B Pawlyk, X Xu, X Liu, JW Bainbridge, M Basche, J McIntosh, HV Tran, A Nathanwi, T Li and RR Ali
Hum Mol Genet . 2009; 18 (12): 2099-114 - Long-term preservation of cones and improvement in visual function following gene therapy in a mouse model of Leber congenital amaurosis (LCA) caused by GC1 deficiency.
M Mihelec, RA Pearson, SJ Robbie, PK Buch, WB Bainbridge, AJ Smith and RR Ali
Human Gene Therapy 2011; 22 (10): 1179-90 - Gene replacement therapy rescues photoreceptor degeneration in a murine model of Leber Congenital Amaurosis lacking RPGRIP.
BS Pawlyk, AJ Smith, PK Buch, M Adamian, D Hong, MA Sandberg, RR Ali, and T Li Invest.
Ophthalmol. Vis. Sci. 2005; 46 (9):3039-3045 - AAV-mediated gene transfer slows photoreceptor loss in the RCS rat model of retinitis pigmentosa.
AJ Smith, FC Schlichtenbrede, M Tschernutter, JW Bainbridge, AJ Thrasher and RR Ali
Mol. Ther. 2003; 8 (2): 118-195 - Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy.
LS Carvalho, J Xu, RA Pearson, AJ Smith, JW Bainbridge, LM Morries, SJ Fliesler, XQ Ding and RR Ali.
Hum Mol Genet. 2011; 20 (16): 3161-75 - Inhibition of retinal neovascularisation by gene transfer of soluble VEGF receptor sFlt-1.
JWB Bainbridge, A Mistry, M De Alwis, E Paleolog, A Baker, AJ Thrasher and RR Ali
Gene Ther. 2002; 9 (5): 320-326 - EIAV vector-mediated delivery of endostatin or angiostatin inhibits angiogenesis and vascular hyperpermeability in experimental CNV.
KS Balaggan, K Binley, M Esapa, RE MacLaren, S Iqball, Y Duran, RA Pearson, O Kan, SE Barker, AJ Smith, JWB Bainbridge, S Naylor and RR Ali
Gene Ther. 2006; 13 (15):1153-1165 - Local administration of an adeno-associated viral vector expressing IL-10 reduces monocyte infiltration and subsequent photoreceptor damage during experimental autoimmune uveitis.
CA Broderick, AJ Smith, KS Balaggan, A Georgiadis, PK Buch, PC Trittibach, SE Barker, G Sarra, AJ Thrasher, AD Dick and RR Ali.
Mol. Ther. 2005; 12 (2): 369-373 - Lentiviral-vector mediated expression of murine IL-1 receptor antagonist or IL-10 reduces the severity of endotoxin-induced uveitis.
P Trittibach, SE Barker, CA Broderick, M Natkunaraja, Y Duran, SJ Robbie, JWB Bainbridge, G-M Sarra, AD Dick and RR Ali
Gene Ther. 2008; 15 (22): 1478-88 - Retinal repair by transplantation of photoreceptor precursors.
RE MacLaren, RA Pearson, A MacNeil, RH Douglas, TE Salt, M Akimoto, A Swaroop, JC Sowden and RR Ali
Nature 2006; 444 (7116): 203-207 - Long term survival of photoreceptors transplanted into the adult murine neural retina requires immune modulation.
EL West, RA Pearson, SE Barker, UF Luhmann, RE MacLaren, AC Barber, Y Duran, AJ Smith, JC Sowden and RR Ali.
Stem Cells. 2010; 28 (11): 1997-2007 - Pharmacological disruption of the outer limiting membrane leads to increased retinal integration of transplanted photoreceptor precursors.
EL West, RA Pearson, M Tschernutter, JC Sowden, RE MacLaren and RR Ali Exp.
Eye Res. 2008; 86 (4): 601-11. - Targeted disruption of outer limiting membrane junctional proteins (Crb1 and ZO-1) increases integration of transplanted photoreceptor precursors into the adult wild-type and degenerating retina.
Pearson RA, Barber AC, West EL, MacLaren RE, Duran Y, Bainbridge JW, Sowden JC, Ali RR.
Cell Transplant. 2010;19 (4):487-503. - Comparative analysis of the retinal potential of embryonic stem cells and amniotic fluid-derived stem cells.
S Decembrini, M Cananzi, S Gualdoni, A Battersby, N Allen, RA Pearson, RR Ali, P De Coppi and JC Sowden
Stem Cells Dev. 2011; 20 (5): 851-63 - Adult ciliary epithelial cells, previously identified as retinal stem cells with potential for retinal repair fail to differentiate into new rod photoreceptors.
Gualdoni S, Baron M, Lakowski J, Decembrini S, Smith A, Pearson R, Ali R, Sowden J.
Stem Cells 2010; 28 (6): 1048-59. - Effective transplantation of photoreceptor precursor cells selected via cell surface antigen expression.
J Lakowski, YT Han, RA Pearson, A Gonzalez-Cordero, EL West, S Gualdoni, AC Barber, M Hubank, RR Ali and JC Sowden
Stem Cells. 2011; 29 (9): 1391-404 - Cone and rod photoreceptor transplantation in models of the childhood retinopathy Leber congenital amaurosis using flow-sorted Crx-positive donor cells.
J Lakowski, M Baron, J Bainbridge, AC Barber, RA Pearson, RR Ali and JC Sowden.
Hum Mol Genet. 2010; 19 (23): 4545-59
Recent Reviews
- Gene supplementation therapy for recessive forms of inherited retinal dystrophies.
Smith AJ, Bainbridge JW, Ali RR.
Gene Therapy (in press) - Induced pluripotent stem cell technology for generating photoreceptors.
Boucherie C, Sowden JC, Ali RR.
Regen Med. 2011 6 (4):469-79. - Regenerative medicine: DIY eye.
Ali RR, Sowden JC.
Nature. 2011 472 (7341):42-3 - Cell transplantation strategies for retinal repair.
West EL, Pearson RA, MacLaren RE, Sowden JC, Ali RR.
Prog Brain Res. 2009;175:3-21.
Professor Robin Ali
Professor of Human Molecular Genetics. BSc, PhD, FMedSci
University College London
Institute of Ophthalmology
Division of Molecular Therapy
11-43 Bath Street
London EC1V 9EL
United Kingdom
Tel: 020 7608 6817
Fax: 020 7608 6991
E-mail:
r.ali[at]ucl.ac.uk